JACS: 硫缺陷硫复铁矿表面水均裂驱动苯乙烯高效水合转化


水(H2O)作为地球上最普遍的环境分子,在维持生命和调控自然过程中发挥重要作用。例如,光系统II通过裂解水释放氧气,塑造了地球富氧大气环境;地球化学过程中H2O衍生的高活性自由基(如•OH和•H)驱动了污染物的自然降解,赋予地表环境自净化的能力。受此启发,科学界致力于发展水分子均裂策略(H2O →•OH + •H),以推动烯烃氢化、环氧化及醇选择性氧化等关键有机转化。然而,水均裂反应面临三重挑战:1)水的闭壳层电子结构赋予其高稳定性,O-H键解离能高达118 kcal mol-1(热力学屏障);2)生成的•OH与•H以极高速率重组(k = 7.0 × 109 mol-1 s-1)(动力学限制);3)低价d区金属配合物虽可借助π*反键轨道活化O-H键,但在氧化还原环境中易结构崩塌,导致自由基重组(材料稳定性)。因此,开发能同步降低O-H键解离能垒并实现自由基空间分离的活性催化材料,成为突破H2O均裂瓶颈的关键路径。
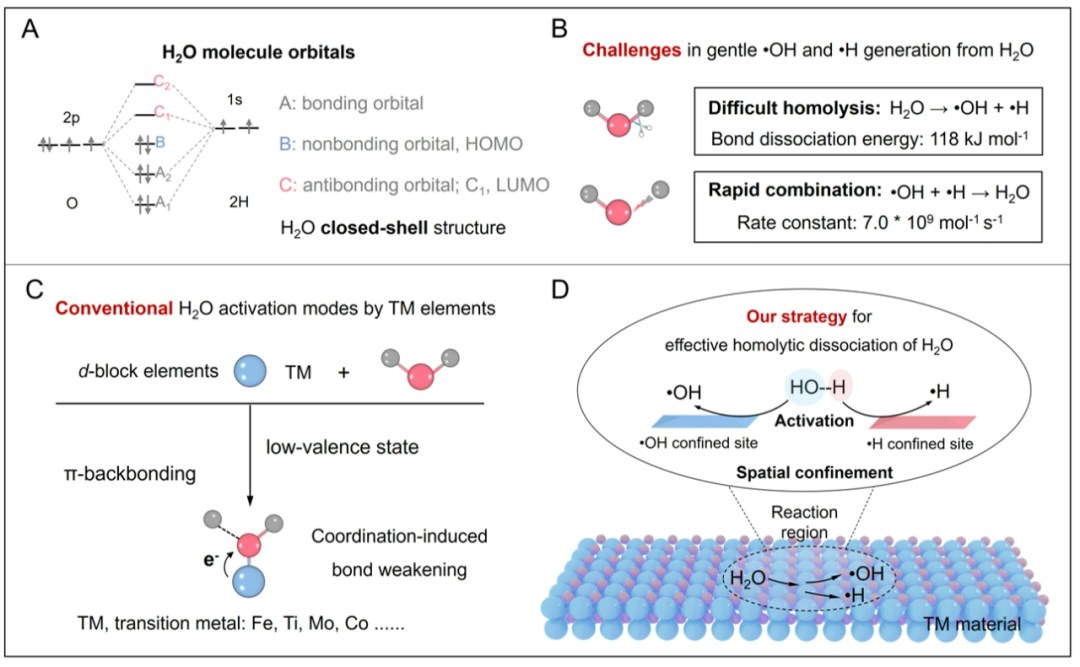
图1、H2O均裂的挑战和机遇。
针对H2O均裂活化面临的挑战,本研究受生物铁硫簇中“Fe活化O-H键/S稳定中间体”协同机制启发,创新设计了含表面单分散硫空位的硫复铁矿(SV-Fe3S4)。其反尖晶石结构中高自旋[Fe3+-Fe2+-Fe3+]构型的反平行排列有利于电子向SV处局域,突破黄铁矿(FeS2)等铁硫矿物的电子传输限制,促进H2O吸附活化并伴随O-H键的解离;同时相邻硫原子成功限域•H并实现•OH/•H空间分离,彻底抑制自由基重组。这种有意思的H2O均裂机制能够在常温常压下驱动苯乙烯水合转化,生成高附加值苯甲醛与甲烷产物,并且整个过程无需额外能量或氧化物摄入,为碳资源-能源联产提供新策略。
SV-Fe3S4表面H2O均裂活化的理论研究
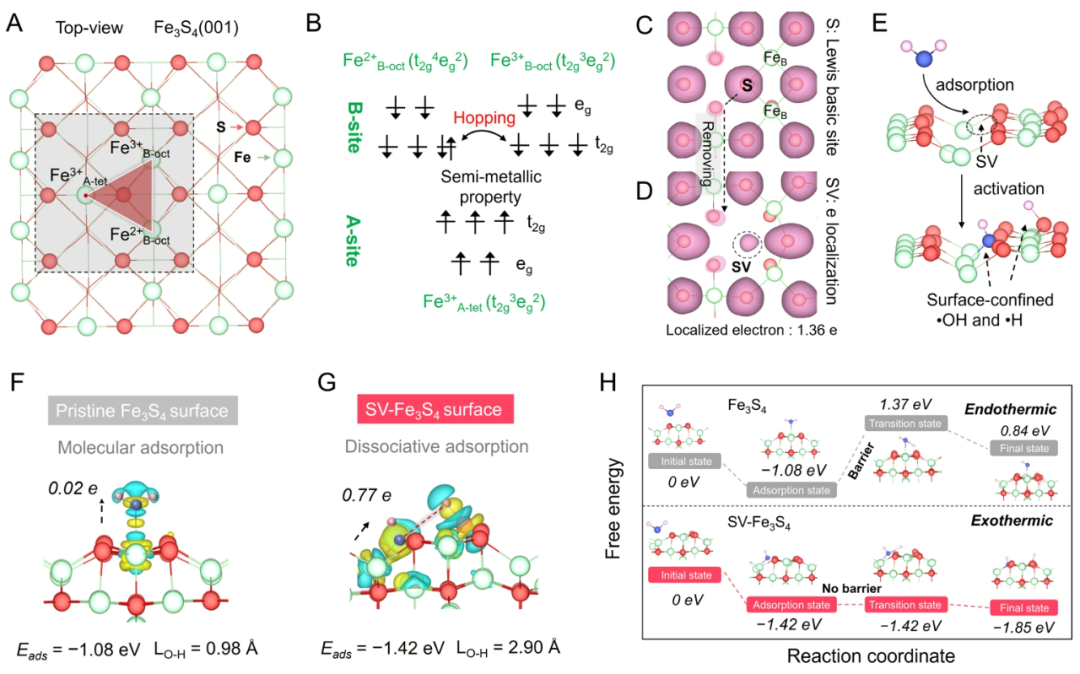
图2、SV-Fe3S4的电子结构特性及其表面H2O吸附活化的理论观点。
理论计算结果表明,Fe3S4具有反尖晶石结构,其高自旋[Fe3+-Fe2+-Fe3+]构型反平行排列促进电子在Fe2+和Fe3+间迁移,有助于体相电子向表面传输。通过在Fe3S4表面构建单分散的硫空位,它既可以作为电子局域中心增强H2O吸附,又通过电子反馈活化将其O-H键从0.97 Å拉伸至2.90 Å;同步发生的均裂解离使•OH在SV周边配位不饱和的FeB位点(≡Fe-•OH)处锚定,而相邻硫原子作为路易斯碱位点可以稳固•H物种(≡S-•H),实现稳定的H2O均裂和•OH/•H空间分离。
硫空位增强H2O与Fe3S4界面相互作用的实验证明
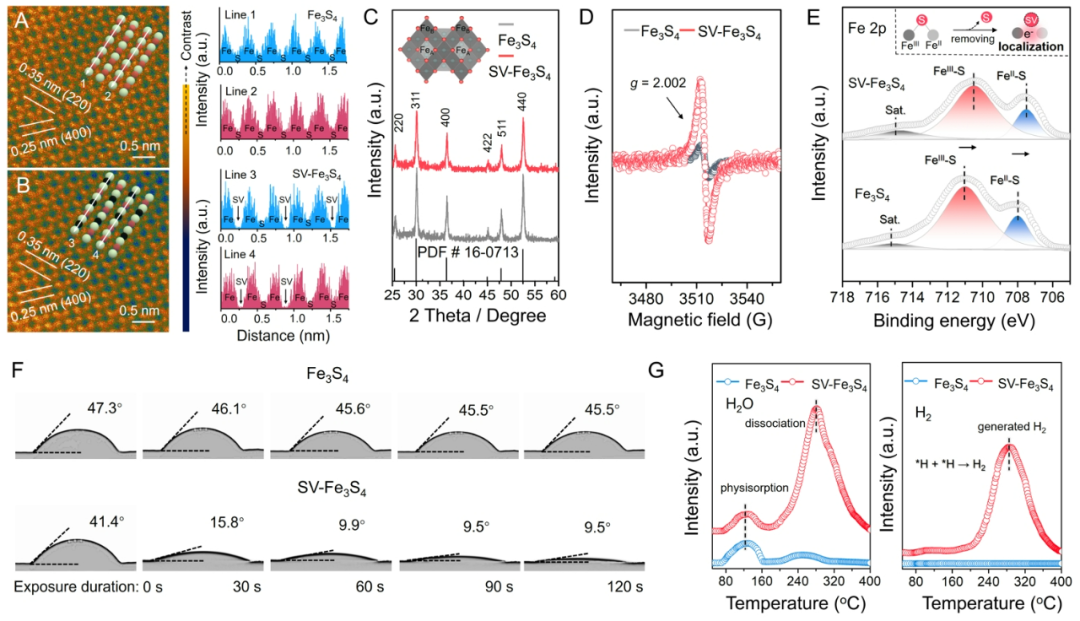
图3、SV-Fe3S4的物相表征及硫空位增强表面H2O吸附活化的实验证据。
采用溶剂热法合成多级花状硫复铁矿(Fe3S4),经真空热处理构建表面单分散硫空位(SV-Fe3S4)。HAADF-STEM表征证实SV-Fe3S4暴露{001}晶面,其硫原子散射强度较原始Fe3S4显著降低,直接可视化表面硫空位。XRD谱显示SV-Fe3S4维持反尖晶石结构(PDF #16-0713),表明表面硫空位构筑并不会改变Fe3S4体相结构。EPR谱中g = 2.002处信号增强及XPS检测到Fe 2p3/2结合能负移,共同印证硫空位处电子局域化引发的表面电荷重新分布,与理论计算结果相符合。水接触角实验揭示:原始Fe3S4表面接触角稳定于45.5°左右,而SV-Fe3S4因硫空位介导的水解离吸附,接触角从41.4°动态降至9.5°。H2O-TPD谱中,SV-Fe3S4在200-360℃出现强化学吸附峰,同步H2-TPD检测到对应H2的脱附峰,为硫空位介导表面H2O均裂提供重要的实验证据。
SV-Fe3S4表面H2O均裂活化的机制探究
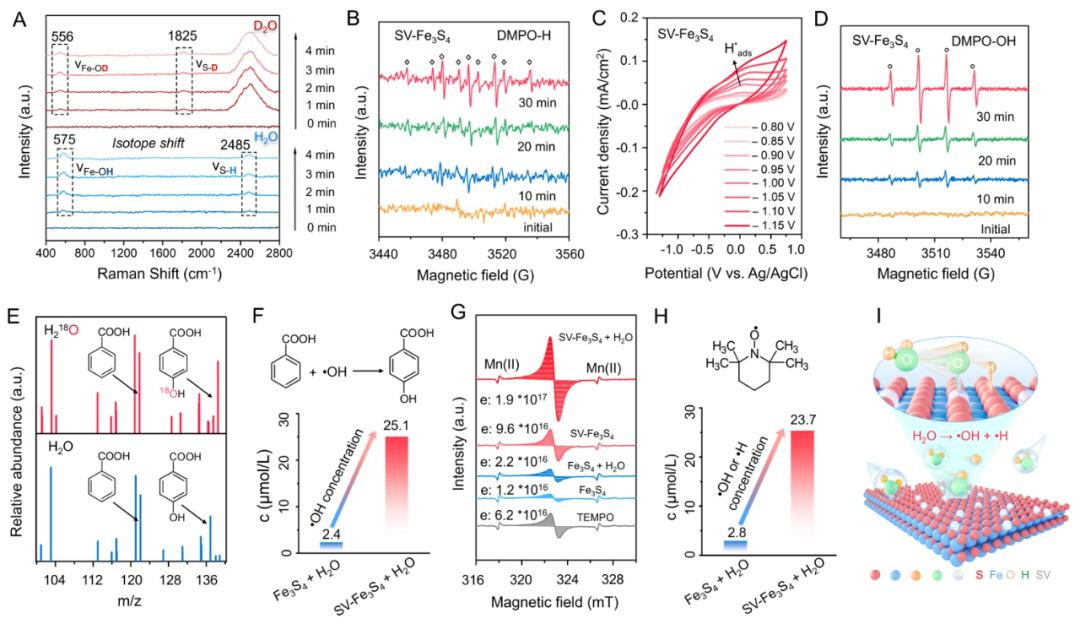
图4、SV-Fe3S4表面H2O均裂的实验证据和机制探究。
In situ Raman在SV-Fe3S4表面575 cm-1和2485 cm-1处分别检测到归属于限域OH(Fe-OH*)和H物种(S-H*)的伸缩振动峰,揭示了H2O均裂过程及解离物种的空间分离特性。以DMPO为自由基捕获剂,通过系列EPR实验证实H2O解离物种以•OH和•H自由基的形式存在。H218O同位素标记实验排除了自由基定性过程中溶解氧的潜在干扰,确认•OH主要来源于H2O解离。采用苯甲酸作为探针分子定量测定表明:SV-Fe3S4-H2O体系•OH浓度达25.1 μmol/L,较原始Fe3S4体系(2.4 μmol/L)提升约10倍。基于TEMPO标准的EPR定量分析显示:SV-Fe3S4-H2O体系中未配对电子浓度的倍增现象符合计量关系—即每个含单未配对电子的硫空位驱动H2O均裂为•OH与•H(各携带一个未配对电子),且计算所得•OH产量与苯甲酸探针法结果一致。上述结果充分证明,SV-Fe3S4表面可实现H2O均裂生成空间限域的•OH和•H自由基。
苯乙烯水合转化
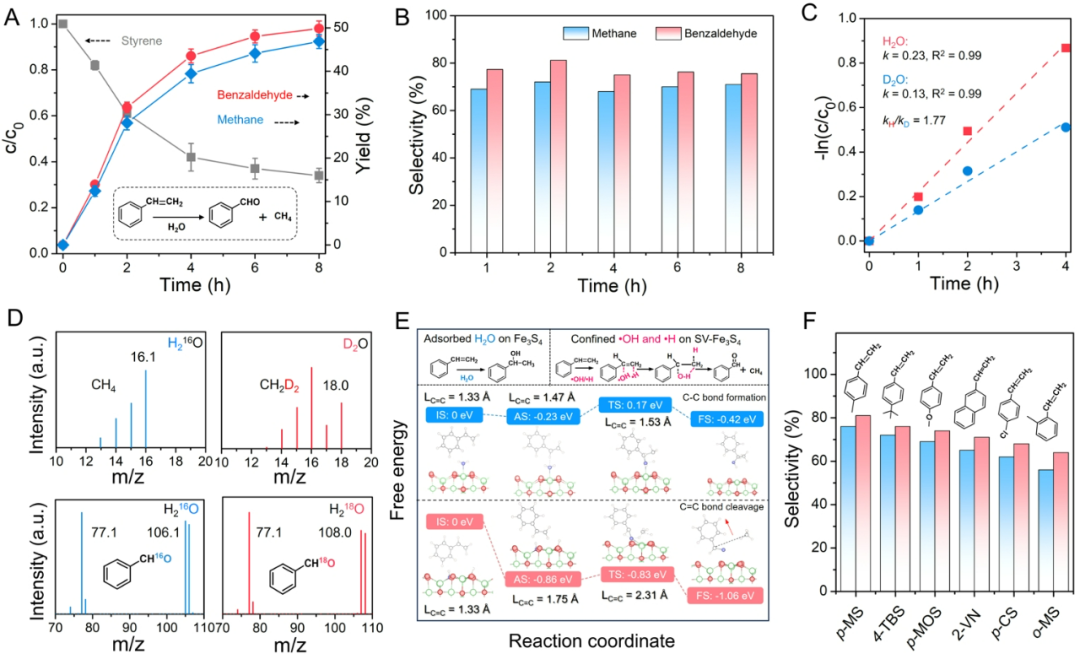
图5、SV-Fe3S4-H2O体系苯乙烯水合转化性能及机理探究。
通过系列苯乙烯水合实验体系,证实SV-Fe3S4可通过H2O均裂生成•OH/•H自由基,进而驱动苯乙烯高效选择性转化为苯甲醛与甲烷。相较于传统酸催化路径(需强酸催化剂经碳正离子中间体生成苯乙醇,成本高且选择性低),SV-Fe3S4在常温常压、纯水为反应媒介条件下,8小时内实现苯乙烯64.6%转化率,苯甲醛与甲烷选择性分别高达80.0%和75.0%,而对照组原始Fe3S4转化苯乙烯效率不足6.0%。关键机理证据包括:1)自由基淬灭实验表明•OH/•H为主要活性物种;2)H218O/D2O同位素标记证实苯甲醛氧原子及甲烷中两个氢原子完全源自H2O解离;3)动力学同位素效应表明O-H均裂为反应的决速步;4)理论计算阐明•OH优先进攻苯乙烯-CH基团形成C-OH键,同时•H加成至-CH2基团,引发C=C键断裂并经分子内重排生成苯甲醛与甲烷。该自由基路径具有普适性,且SV-Fe3S4催化剂可循环使用,整个体系无需外加试剂或能量输入,为碳资源高值化转化提供了新策略。
本研究首次证实具有反尖晶石结构的SV-Fe3S4可突破常温下H2O稳定均裂热力学和动力学壁垒。机制上,硫空位通过高自旋Fe反平行排列实现电子局域化,驱动O-H键无势垒解离成表面•OH与•H;相邻硫原子凭借强路易斯碱性稳定•H并促进空间分离。得益于此,SV-Fe3S4-H2O体系成功实现了苯乙烯及其衍生物高选择性转化为高附加值苯甲醛与富能甲烷产物。此外,该H2O均裂机制有望开辟新范式:1)在有机合成领域,拓展至更多不饱和化合物的C=C/C≡C键高值化转化,设计串联反应提升原子经济性;2)在环境治理方面,利用同步产生的•OH/•H实现污染物深度氧化及生活用水高效消毒,避免传统工艺的化学添加剂残留。
转自https://mp.weixin.qq.com/s/aAmt9wMzZAVpoizY8aML6A