上海交通大学张礼知/李浩/李美琪,最新Nature子刊!分子氧和黄铁矿合成高自旋表面Feᴵⱽ=O以实现甲烷选择性氧化!凌灿灿一作
受自然界启发的高自旋Feᴵⱽ=O物种合成策略实现了温和条件下甲烷高效氧化。
2025年8月16日,上海交通大学张礼知(国家杰青/长江学者)、李浩、李美琪、凌灿灿团队在Nature Communications期刊发表题为“High-spin surface Feᴵⱽ=O synthesis with molecular oxygen and pyrite for selective methane oxidation”的研究论文,凌灿灿为论文第一作者,李美琪、李浩、张礼知为论文共同通讯作者。

DOI: 10.1038/s41467-025-63087-w
该研究通过在黄铁矿(FeS₂)表面构建硫桥连的双=Feᴵᴵ...Feᴵᴵ位点,模拟可溶性甲烷单加氧酶(sMMO),成功在室温和常压下驱动O₂形成高自旋(S=2)表面Feᴵⱽ=O物种。策略性移除桥接S原子创造了活性位点,通过瞬态≡Fe-O-O-Fe≡中间体促进O₂活化,实现O-O键均裂。所得Feᴵⱽ=O具有不对称扭曲配位环境,降低了晶体场分裂能,有利于未成对电子占据高能d轨道。该构型可通过氧转移反应将CH₄高效转化为CH₃OH,合成效率达TOF=27.4 h⁻¹,选择性为87.0%,性能优于大多数常温O₂驱动体系,甚至超越多种H₂O₂介导体系。该工作为合成高自旋表面Feᴵⱽ=O提供了新方法,并揭示了金属自旋态调控在非酶促甲烷活化中的关键作用。
甲烷(CH₄)作为天然气的主要组分,其高效利用既能作为能源密集型燃料和大宗化学品的原料,又可缓解其温室效应。当前工业策略通过将CH₄重整为合成气(CO与H₂混合物),再经150–300°C温度范围和1–5 MPa压力条件下的费托过程制备液态烃(如甲醇CH₃OH)。然而,此类热驱动路线资本密集且依赖基础设施,难以适用于小型设施和海上钻井平台或页岩气田等分散区域,导致CH₄运输的经济性问题常以燃烧放空处理。因此,温和条件下CH₄选择性氧化被视为化学领域的"圣杯"。但CH₄的非极性四面体结构赋予其高达439.3 kJ mol⁻¹的C–H键解离能,显著阻碍其活化过程。故通常需采用强效且昂贵的氧化剂(如发烟硫酸、一氧化二氮、过氧化氢或臭氧),通过均相或负载型过渡金属催化中心实现CH₄活化。
分子氧(O₂)是CH₄活化最绿色经济的氧化剂,为CH₄升级提供高性价比替代方案。但三线态O₂与单线态CH₄间的直接反应存在自旋禁阻特性,需超过100°C的反应温度克服自旋态失配以促进反应。值得注意的是,自然界通过金属酶(如可溶性甲烷单加氧酶sMMO)在常温下实现CH₄高效转化为CH₃OH:该酶利用双铁(II)中心活化O₂,促进O–O键均裂并生成高价铁氧(Feᴵⱽ=O)物种。尽管已投入大量努力合成非血红素双铁(II)配合物或在沸石/金属有机框架上固载双Feᴵᴵ中心以模拟sMMO功能,这些人造体系使用O₂作为终端氧化剂时,常生成中自旋态Feᴵⱽ=O,其反应活性显著低于酶促生成的高自旋Feᴵⱽ=O物种。
黄铁矿作为储量丰富、无毒廉价的含硫矿物(化学式FeS₂),广泛应用于环境治理、电化学与光伏领域。其表面具有丰富的5配位≡Feᴵᴵ(FeS₂表面Feᴵᴵ)通过硫原子桥连(≡Feᴵᴵ–S–Feᴵᴵ≡),成为理想的O₂活化双≡Feᴵᴵ载体。但FeS₂表面直接解离O₂形成高自旋Feᴵⱽ=O面临空间与能量双重挑战:首先,桥接硫原子的大原子半径会空间位阻O₂在≡Feᴵᴵ–S–Feᴵᴵ≡位点的吸附;其次,成键硫原子产生的强配体场增大Fe的d轨道能隙,导致晶体场过度分裂,非预期地形成具有紧密电子对的低自旋≡Feᴵᴵ。这种电子构型进一步阻碍高效电子转移——该过程对裂解高能垒O–O键及后续高自旋Feᴵⱽ=O形成至关重要。解决这些挑战需精准设计双≡Feᴵᴵ位点的几何结构并精细调控FeS₂的配体场性质,这对实现温和条件下高效O₂解离及高自旋Feᴵⱽ=O生成具有决定性意义。
该研究通过在FeS₂(001)表面移除桥接S原子构建双≡Feᴵᴵ…Feᴵᴵ≡位点,可实现O₂均裂生成高自旋表面Feᴵⱽ=O,从而功能模拟sMMO的O₂活化策略。研究采用综合理论计算与先进显微表征手段,系统探究硫空位如何影响O₂吸附活化行为、双≡Feᴵᴵ…Feᴵᴵ≡位点的晶体场对称性及高自旋Feᴵⱽ=O形成过程。通过评估室温常压下生成的高自旋Feᴵⱽ=O以O₂为氧化剂选择性氧化CH₄至CH₃OH的反应性能,旨在开发高效的非酶促CH₄选择性转化方法。
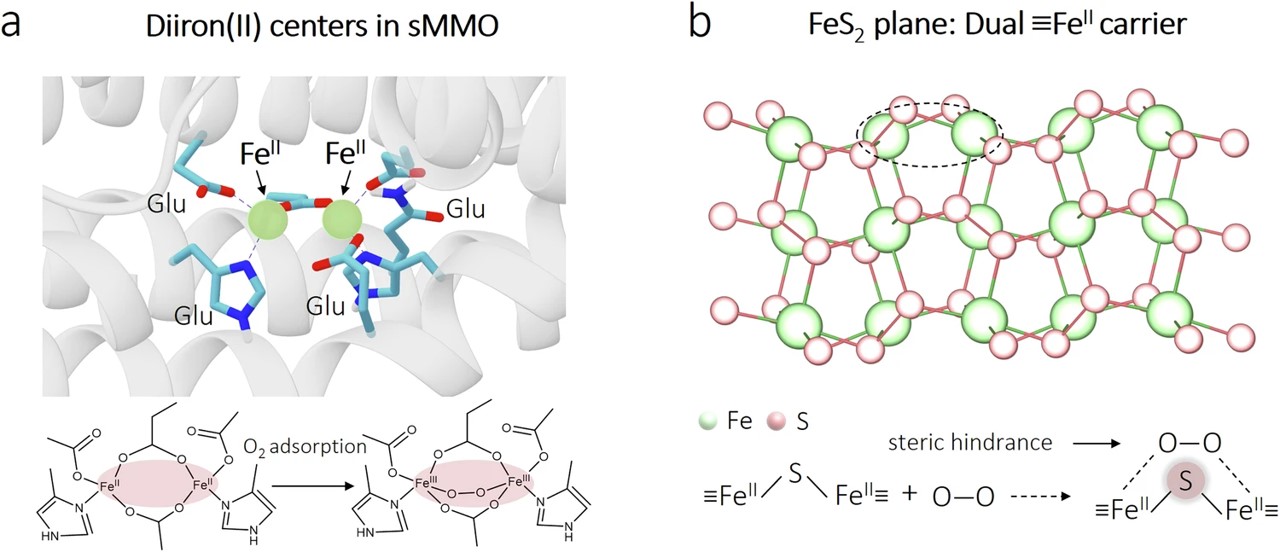
图1| 双核铁中心氧吸附机制。(a) 可溶性甲烷单加氧酶(sMMO,蛋白质数据库1MTY)羟基酶组分中双铁(II)中心吸附O₂示意图。(b) FeS₂平面上双≡Feᴵᴵ(≡表示材料表面)分布,展示桥接S原子对O₂吸附的空间排斥作用。
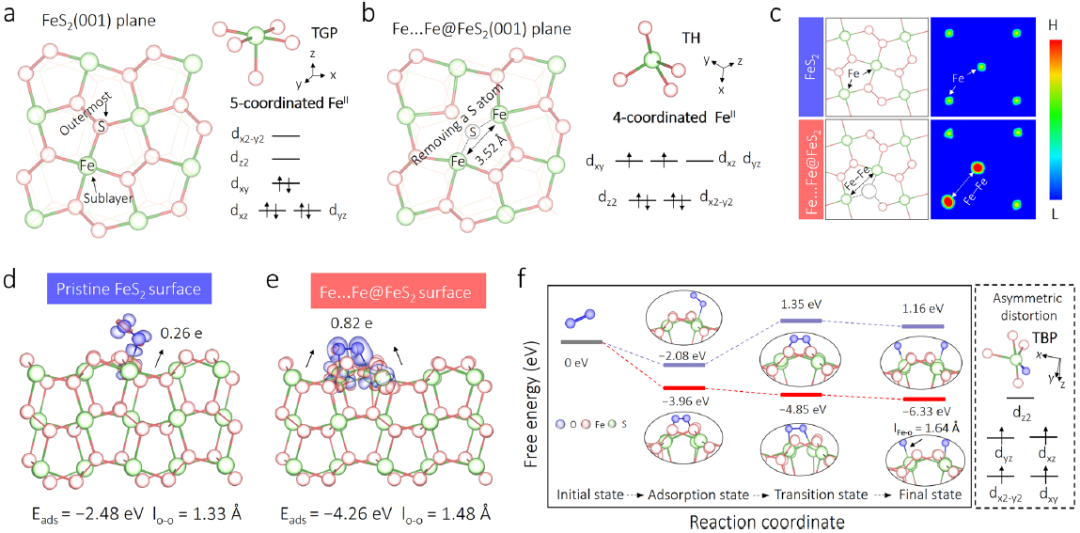
图2| O₂活化和表面高自旋Feᴵⱽ=O合成的理论解析。(a) FeS₂(001)表面5配位≡Feᴵᴵ的电子分布(四方锥几何构型)。(b) 移除桥接硫原子后双≡Feᴵᴵ...Feᴵᴵ位点的电子分布(四面体几何构型)。(c) FeS₂和Fe...Fe@FeS₂优化结构及二维自旋密度图(L:低自旋强度,H:高自旋强度)。(d) O₂在原始FeS₂表面5配位≡Feᴵᴵ位的端基吸附。(e) O₂在Fe...Fe@FeS₂表面双4配位≡Feᴵᴵ...Feᴵᴵ位的桥连吸附(附差分电荷密度图)。(f) FeS₂和Fe...Fe@FeS₂表面O₂解离及Feᴵⱽ=O形成的自由能变化(插图为表面Feᴵⱽ=O的三角双锥几何d电子分布)。
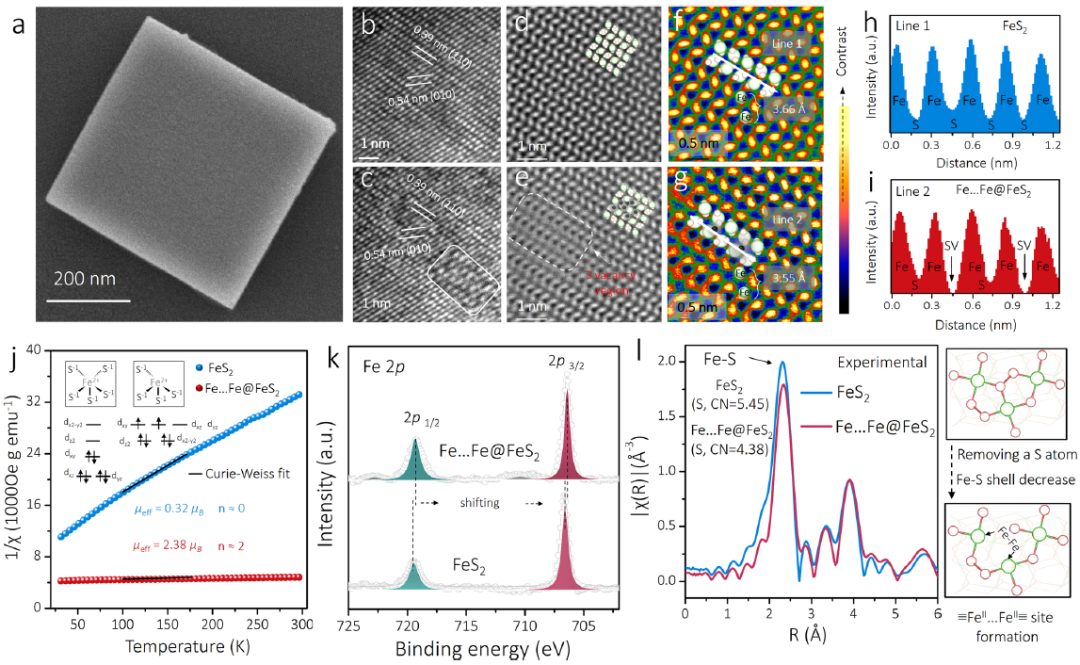
图3 | Fe...Fe@FeS₂的综合表征。(a) 样品SEM图像。(b,c) FeS₂和Fe...Fe@FeS₂的HRTEM图像(白色矩形框标示模糊区域)。(d,e) FeS₂和Fe...Fe@FeS₂的HAADF-STEM图像(插图为理论模型,黑色表示硫空位)。(f,g) 对应伪彩色HAADF-STEM图像。(h,i) 图f,g白色箭头区原子强度线扫描。(j) FeS₂和Fe...Fe@FeS₂的1/χ–温度曲线(含μeff计算值)。(k) Fe 2p XPS谱。(l) Fe扩展XAFS谱傅里叶变换及双≡Feᴵᴵ...Feᴵᴵ位点形成示意图。
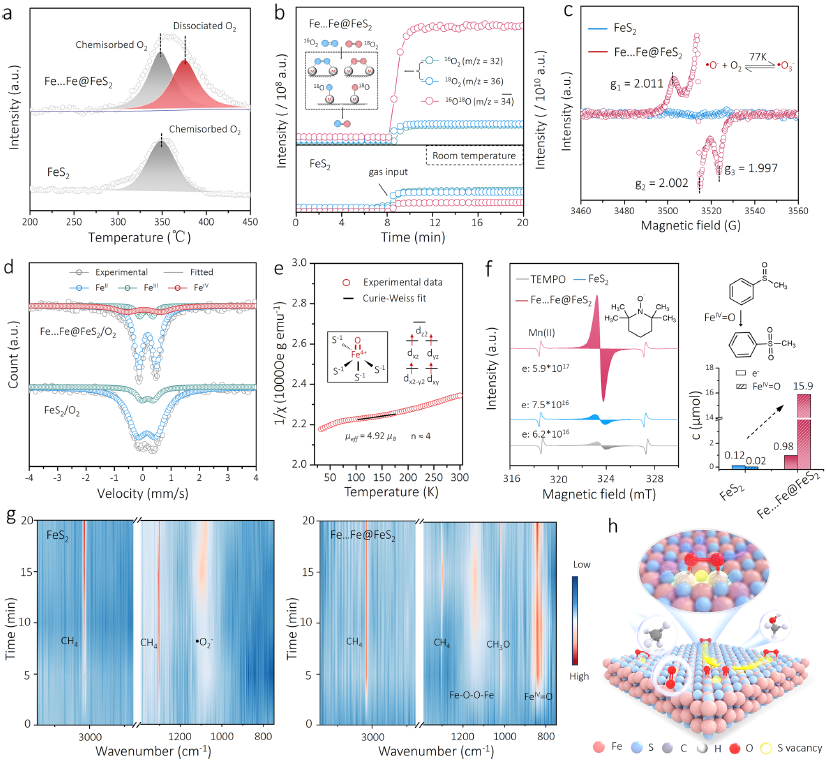
图4 | 高自旋Feᴵⱽ=O活化甲烷的机理研究。(a) FeS₂和Fe...Fe@FeS₂的O₂-TPD谱。(b) 氧同位素交换质谱图及双≡Feᴵᴵ...Feᴵᴵ位点O₂解离/重组示意图。(c) Fe...Fe@FeS₂上O₂活化/解离的低温EPR谱。(d) O₂处理后样品的⁵⁷Fe穆斯堡尔谱(实验值与拟合值)。(e) O₂处理样品的1/χ–温度曲线(含μeff计算值)。(f) EPR定量局域电子浓度及PMSO捕获的Feᴵⱽ=O生成量。(g) CH₄和O₂在样品表面吸附的原位FTIR谱。(h) 双≡Feᴵᴵ...Feᴵᴵ位点高自旋Feᴵⱽ=O选择性氧化CH₄示意图。
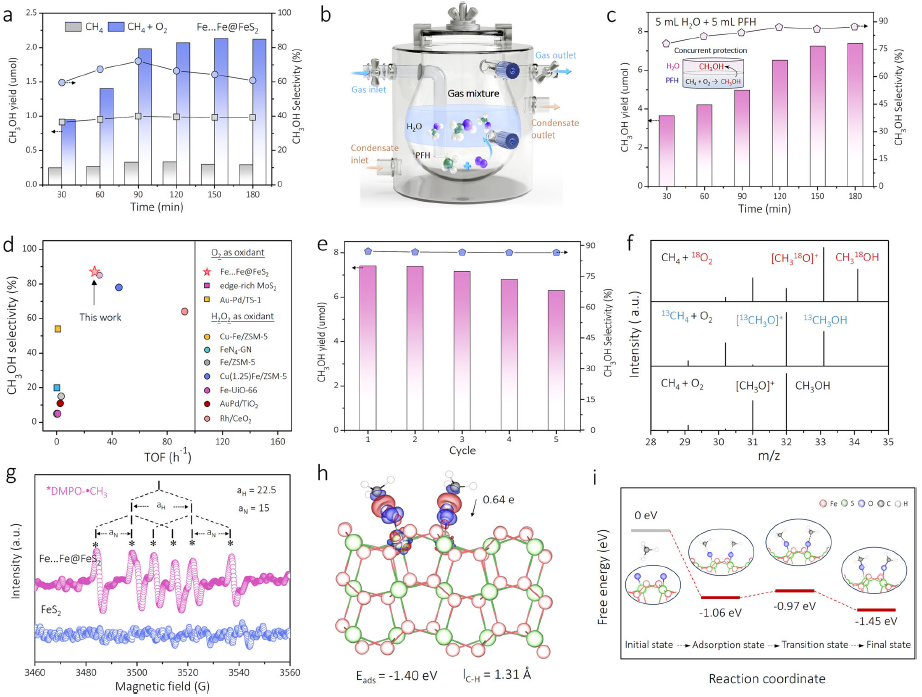
图5| CH₄选择性氧化制CH₃OH。(a) Fe...Fe@FeS₂体系液相产物分析。(b) 全氟己烷(PFH)/水双溶剂反应器示意图。(c) 双溶剂体系CH₃OH产率/选择性。(d) Fe...Fe@FeS₂/O₂体系与其他气-液-固三相催化体系的TOF/选择性对比。(e) PFH/水体系中CH₄氧化的循环稳定性。(f) 同位素标记实验产物GC-MS分析。(g) DMPO-·CH₃加合物EPR谱。(h) 高自旋Feᴵⱽ=O吸附CH₄的理论模型(附差分电荷密度图)。(i) CH₄活化/氧化的自由能变化曲线。
总之,该研究证明,通过在黄铁矿表面构建双≡Feᴵᴵ...Feᴵᴵ≡位点,以O₂为氧化剂可合成具有酶促反应活性的高自旋Feᴵⱽ=O物种。移除桥接硫原子形成的双位点通过桥连=Fe-O-O-Fe=中间体与O₂的π*反键轨道有效作用,促进O-O键均裂生成高自旋表面Feᴵⱽ=O。其不对称扭曲配位环境降低晶体场分裂能,促使未成对电子占据高能d轨道。该高自旋表面Feᴵⱽ=O通过氧转移反应将CH₄高效转化为CH₃OH(TOF=27.4 h⁻¹,选择性87.0%),性能超越多数环境O₂驱动体系及H₂O₂介导体系。该工作为高自旋表面Feᴵⱽ=O合成提供了新策略,并揭示了金属自旋态调控在非酶促甲烷活化中的重要性。
转自https://mp.weixin.qq.com/s/h7HBXDjrxs4ubQ6ZXO1a9Q