别再纠结中间自旋了!张礼知教授等团队NC 报道硫桥“断链”神操作,解锁高活性高自旋Fe(IV)=O,室温下O₂活化甲烷不是梦!

期刊信息

2025年8月16日,上海交通大学凌灿灿、李美琪、李浩、张礼知团队与华中师范大学赵进才团队合作在Nature Communications期刊发表题为“High-spin surface FeIV = O synthesis with molecular oxygen and pyrite for selective methane oxidation”的研究论文,上海交通大学凌灿灿为论文第一作者,李美琪、李浩、张礼知为论文共同通讯作者。
广泛背景 → 甲烷(CH₄)选择性氧化制甲醇是化学领域的“圣杯”反应,但条件苛刻。
核心问题 → 以O₂为氧化剂在温和条件下活化CH₄存在自旋禁阻难题,难以形成高活性的高价铁氧物种。
现有方法及其局限 → 自然界中的甲烷单加氧酶(sMMO)通过双铁中心形成高自旋Fe(IV)=O物种解决此问题,但人工模拟体系使用O₂时,往往生成活性较低的中、低自旋物种。
聚焦新策略 → 选用储量丰富、廉价的黄铁矿(FeS₂)作为模型,通过移除其表面的桥联硫原子,构建独特的双Fe(II)活性位点(≡FeII…FeII≡)。
阐述假设的机制链条 → 移除硫桥 → 形成配位不饱和、扭曲的双铁位点 → 铁中心电子结构改变(低自旋→中自旋)→ 促进O₂以桥联模式吸附(≡Fe−O−O−Fe≡)并发生O-O键均裂 → 生成的Fe(IV)=O物种因其不对称配位环境而稳定在更高活性的高自旋态(S=2)→ 高自旋Fe(IV)=O通过氢提取/氧反弹机理高效氧化甲烷。
预告研究成果 → 在室温常压下实现了高效、高选择性的甲烷到甲醇的转化,性能优于大多数O₂驱动体系,甚至超越许多H₂O₂体系。
让我们来看看如何对这篇文献深刻的拆解,清晰的呈现到你面前,制作不易,方便的话大家可以点赞转发哟(公众号加上星标,不迷路哟),尤其建议刚入门的研究生同学可以对着文献和我们的解析进行对比阅读,有利于打开大家的思路,理清逻辑思路。
话不多说,今天的这篇文章解决的是一个公认的“圣杯级”难题——温和条件下用氧气直接把甲烷变甲醇。这个反应的难点在于,三重态的氧气和单重态的甲烷“自旋禁阻”,反应不来。大自然用甲烷单加氧酶(sMMO)里的高自旋Fe(IV)=O物种解决了这个问题,但人工模拟却屡屡碰壁,造出的铁氧物种总是“自旋不对”,活性差一大截。那么,作者是如何在这样一个“硬骨头”上取得突破的呢?这就引出了本文最精妙的地方:选材和改造的“巧”。他们没有选择传统的高端氧化物或MOF载体,而是把目光投向了地球上随处可见、甚至有点“土”的黄铁矿(FeS₂)。这本身就是一种逆向思维。
更核心的创新在于,他们对黄铁矿动了一次精准的“微创手术”——不是常规地引入氧空位或掺杂,而是通过真空退火,精准地移除连接两个表面铁原子的“硫桥”。这一步操作看似简单,实则一箭三雕:
1. 空间解锁:拆掉硫桥,为O₂分子的吸附和活化腾出了宝贵的空间。
2. 电子重构:打破了原有的配位场对称性,使Fe(II)的电子排布从“懒惰”的低自旋态,转变为更“活跃”的中自旋态。
3. 协同活化:构筑出的“肩并肩”的双铁位点(≡FeII…FeII≡)完美地模拟了sMMO,能够以桥联方式“夹住”O₂分子,极大地促进了O-O键的断裂。
最关键的是,这一系列操作的最终落脚点,是成功地在表面原位生成并稳定了梦寐以求的高自旋(S=2)Fe(IV)=O物种。这正是解锁甲烷C-H键活性的“金钥匙”。整个逻辑链条从“移除一个硫原子”这个简单的宏观操作,层层递进,最终追溯到d轨道电子排布和自旋态这个最本质的微观根源,论证清晰,堪称典范。这篇文章完美诠释了如何通过对廉价材料进行精巧的结构工程,来模拟复杂的生物酶功能,从而攻克基础化学中的重大挑战。
我们再看这篇文章的DFT理论计算部分(主要集中在Figure 2),作者团队的操作堪称“理论先行”的典范,它不是实验的附庸,而是整个研究的“设计蓝图”和“导航系统”!
首先,模型构建精准且具有前瞻性。作者没有随意抓取一个FeS₂的表面,而是聚焦于其热力学最稳定的(001)面。更关键的是,他们精准地模拟了实验中将要进行的操作——移除一个桥联的硫原子,从而构建了含有硫缺陷的“双铁位点(dual ≡FeII…FeII≡)”模型(图2b)。这使得整个理论计算从一开始就瞄准了核心的科学问题:一个简单的硫缺陷,究竟能给FeS₂的催化性能带来怎样翻天覆地的变化?
其次,计算逻辑层层递进,直击要害。整个计算过程围绕着一个核心主线展开:硫缺陷如何调控铁中心的电子自旋态,并最终实现对O₂分子的“无障碍”活化?
1. 揭示“原始问题”(图2a):作者首先计算了完美FeS₂(001)表面的状态。结果表明,其表面的五配位Fe(II)位点,由于受到周围硫原子强大的配体场作用,导致Fe的d轨道发生巨大的晶体场分裂,电子倾向于成对排布,形成低自旋(S=0)状态。这种状态电子“惰性”十足,是其无法有效活化O₂的根本原因。
2. 提出“解决方案”(图2b):通过在模型中移除一个桥联S原子,作者发现,Fe中心的配位数从5降为4,局部对称性被打破,几何构型从四方锥(TGP)变为扭曲的四面体(TH)。这一“微创手术”直接导致了电子排布的重构,产生了两个未成对电子,使Fe(II)位点转变为中间自旋(S=1)状态。
3. 验证“电子调控”效果(图2c):通过计算自旋密度图,作者直观地展示了这一变化。缺陷位点(Fe…Fe@FeS₂)的Fe中心呈现出明显更强且不对称的自旋密度(图中红色区域),完美印证了从低自旋到中间自旋的转变。这为后续的O₂活化提供了必要的“电子灵活性”。
4. 阐明“活化机理”(图2d, 2e):这是最精彩的部分。作者对比了O₂在两种表面的吸附和活化行为。
在完美表面(图2d),O₂只能以“端基”方式吸附,活化效果微弱(O-O键仅拉长至1.33 Å)。
而在缺陷表面(图2e),两个相邻的、处于中间自旋态的Fe位点协同作用,使O₂能够以“桥联”模式(≡Fe−O−O−Fe≡)被捕获。这种模式极大地促进了从双铁中心到O₂反键轨道的电子转移(高达0.82 e),导致O-O键被急剧削弱并拉长至1.48 Å。
5. 预测“终极产物”(图2f):基于上述强大的活化,作者计算了O-O键断裂的反应路径。结果令人震撼:在缺陷表面上,O-O键的断裂是一个自发的、无能垒的过程!而在完美表面,能垒则高达3.43 eV,在室温下绝无可能发生。更重要的是,计算预测了断裂后形成的产物正是在甲烷氧化中扮演关键角色的高自旋(S=2)FeIV=O物种。其扭曲的三角双锥(TBP)几何构型减小了晶体场分裂,使得电子能够占据更高能量的d轨道,从而形成高反应活性的高自旋态。
总而言之,这篇论文的DFT计算部分,从“识别问题”(低自旋FeII)出发,通过“结构工程”(制造硫缺陷),实现了“电子调控”(自旋态转变),最终阐明了“机理路径”(桥式吸附与无垒断裂),并精准预测了“关键活性物种”(高自旋FeIV=O)。整个过程如同一场精密的战术推演,为后续的实验合成与性能验证提供了无懈可击的理论指导。
摘要是文章的“精华浓缩版”,咱们按照“背景-问题-方案-亮点-意义”的框架来解析:
核心亮点与成果:
研究意义:
这项研究不仅提供了一种简单易行的方法来合成高活性的高自旋FeIV=O物种,更重要的是,它揭示了通过“调控金属自旋态”来设计高效非酶催化剂的巨大潜力,为温和条件下的甲烷活化开辟了新思路。
本研究的创新解决之处在于,巧妙地利用地球上储量丰富的黄铁矿(FeS₂),通过简单的真空退火方法制造出表面硫缺陷,从而构建出一种能模拟天然酶活性中心的“双铁位点”。该位点的核心功能是调控铁中心的电子自旋态,实现了从“惰性”低自旋到“活性”高自旋FeIV=O物种的转变,最终攻克了在温和条件下用O₂高效选择性氧化甲烷的难题。
证据一(结构确认):我们真的造出了“双铁位点”吗?
HAADF-STEM(图3d-g):高分辨电镜给出了最直观的原子级证据。与完美的FeS₂晶格相比,处理后的Fe…Fe@FeS₂表面出现了明显的硫原子“空洞”(dark spots的fading),并且测得的Fe-Fe原子间距(3.55 Å)与理论模型(3.52 Å)高度吻合,直接证实了桥联硫原子的成功移除和双铁位点的形成。
EXAFS(图3l):X射线吸收谱进一步从配位环境上提供了定量证据。Fe…Fe@FeS₂中Fe-S的配位数(CN ≈ 4.38)显著低于完美FeS₂(CN ≈ 5.45),这与五配位Fe转变为四配位Fe的理论模型完全一致。
证据二(电子态验证):铁中心的自旋态真的改变了吗?
磁化率测试(图3j):这是验证自旋态变化的关键。测试表明,Fe…Fe@FeS₂的有效磁矩(μeff = 2.38 μB)远高于原始FeS₂(0.32 μB),计算出的未成对电子数从0增加到了2。这有力地证实了Fe(II)从低自旋(S=0)成功转变为理论预测的中间自旋(S=1)状态。
证据三(机械性能的革命性提升):
O₂同位素交换(图4b):实验表明,Fe…Fe@FeS₂能极大地促进¹⁶O₂和¹⁸O₂的交换反应,生成¹⁶O¹⁸O,证明其表面具有强大的O-O键断裂能力。
⁵⁷Fe穆斯堡尔谱(图4d):这是鉴定FeIV=O物种及其自旋态的“王牌证据”。在通入O₂后,只有Fe…Fe@FeS₂的谱图中出现了一个全新的、归属于FeIV物种的信号峰。其同质异能位移和四极分裂参数与文献报道的高自旋(S=2)FeIV=O物种的特征值高度吻合,为关键活性物种的生成提供了决定性证据。
证据四(反应机理追踪):是FeIV=O在氧化甲烷吗?
原位FTIR光谱(图4g):在CH₄和O₂共存的条件下,清晰地观测到FeIV=O的特征峰(840 cm⁻¹)出现,随后,表面甲氧基(OCH₃)的特征峰(1020 cm⁻¹)生成。这动态地展示了FeIV=O物种将甲烷氧化为甲醇中间体的过程。
同位素标记实验(图5f):通过使用¹³CH₄和¹⁸O₂,质谱结果显示产物分别为¹³CH₃OH和CH₃¹⁸OH。这确凿无疑地证明了产物甲醇中的碳原子来自甲烷,氧原子来自O₂分子。
EPR自旋捕获(图5g):实验中成功捕获到了DMPO-•CH₃加合物信号,这是甲烷C-H键被断裂后生成甲基自由基(•CH₃)的直接证据,与理论计算提出的“氢提取/氧反弹”机理完全契合。
前言解析
-
前言部分,作者为我们描绘了一幅清晰的研究“路线图”:
开篇点题,直指化学领域的“圣杯”难题——在温和条件下选择性氧化甲烷。作者首先点明了现有工业路线(重整为合成气)高能耗、高成本的弊端,从而引出了使用分子氧(O2)直接氧化甲烷的理想前景。接着,作者阐述了此路线的核心障碍——三重态O2与单重态CH4之间的自旋禁阻反应,这使得反应通常需要高温。话锋一转,作者将目光投向自然界的精妙设计——可溶性甲烷单加氧酶(sMMO)。该酶利用其双铁(II)中心激活O2,生成高反应活性的高自旋FeIV=O物种,从而在常温下高效催化甲烷氧化。这为人工模拟提供了绝佳范本。然而,作者指出现有的人工模拟体系(如非血红素配合物或负载型双铁中心)在使用O2作氧化剂时,往往生成反应性较低的“中自旋”FeIV=O,无法媲美天然酶的效率。
作者终于亮出了自己的创意!他们选择了一种储量丰富、无毒且廉价的硫基矿物——黄铁矿(FeS2)。黄铁矿表面天然存在着由硫原子桥联的双FeII位点(≡FeII-S-FeII≡),这使其成为承载双铁中心的理想载体。但作者敏锐地指出了其内在缺陷:第一,桥联硫原子较大的原子半径会产生空间位阻,阻碍O2的吸附;第二,硫原子提供的强配体场效应导致FeII处于“低自旋”状态,不利于O2的活化。
最后,作者清晰地阐述了他们的核心论点和解决方案:“Herein, we demonstrate...”。通过精确地移除桥联的硫原子来构建“无桥联的双≡FeII…FeII≡位点”。这种策略旨在通过改变铁中心的几何结构和配体场环境,调控其电子自旋态,从而促进O2的解离并生成高活性的“高自旋”表面FeIV=O物种,最终实现对sMMO功能的有效模拟和高效的甲烷选择性氧化。

图1 概念引入:模拟天然酶和黄铁矿的机遇与挑战
-
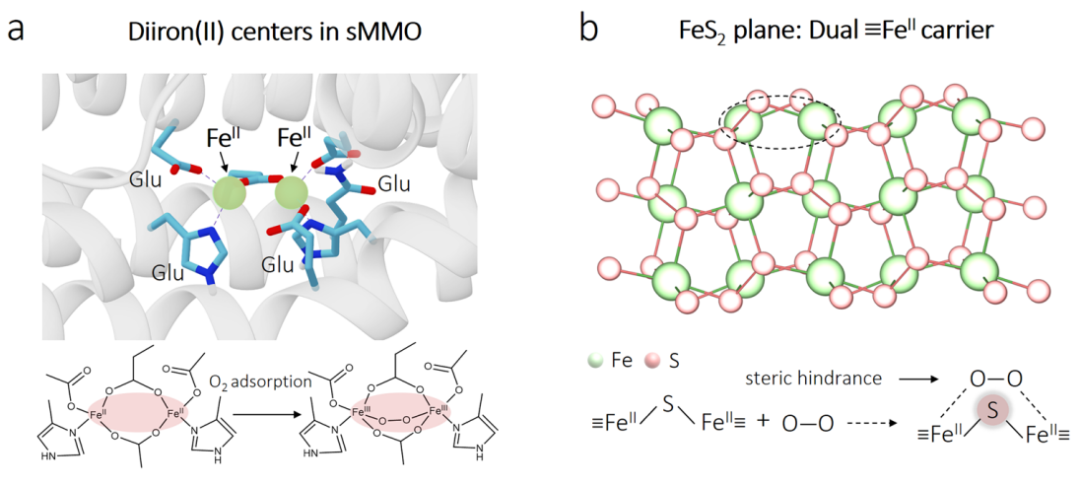
-
(a) 展示了天然sMMO酶中用于激活O2的双铁中心结构,作为本研究的灵感来源。
(b) 展示了黄铁矿(FeS2)表面的双FeII位点分布,并形象地指出了桥联的硫原子对O2分子的空间排斥作用,揭示了天然黄铁矿难以激活O2的原因。
图2 理论计算:揭示硫空位如何激活O2并生成高自旋FeIV=O
-
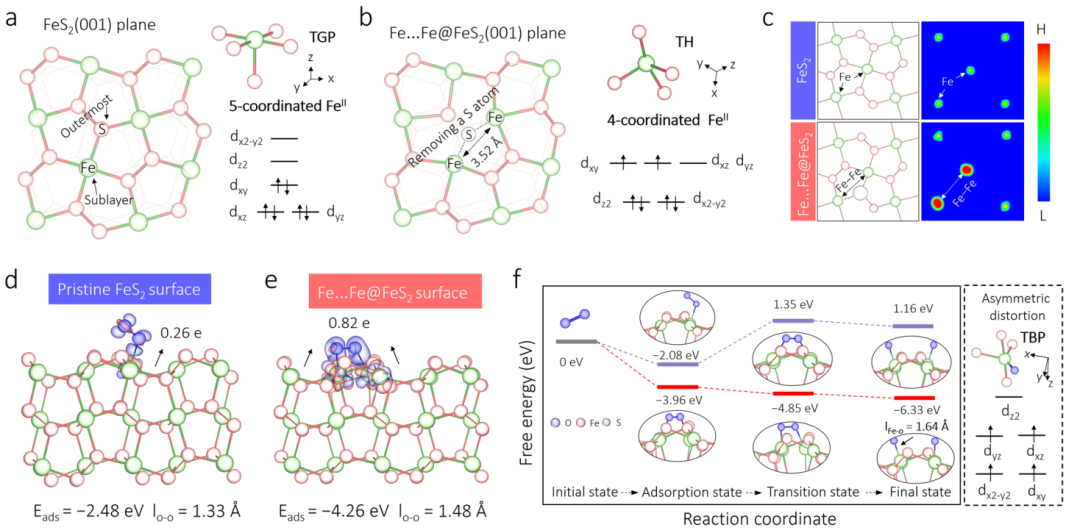
-
(a) 和 (b) 对比了原始FeS2表面和移除桥联硫原子后(Fe…Fe@FeS2)的结构与电子排布。结果显示,硫的移除使Fe的配位数从5变为4,几何构型从四方锥变为扭曲的四面体,电子自旋态从低自旋(S=0)转变为中自旋(S=1)。
(c) 自旋密度图直观地证实了硫空位处的Fe位点具有更高的自旋密度和不对称性。
(d) 和 (e) 对比了O2在两种表面的吸附模式。在工程化的双Fe位点上,O2以“桥联模式”(≡Fe−O−O−Fe≡)被强力吸附和活化,O-O键被显著拉长。
(f) 反应路径的自由能图揭示了关键结论:在工程化的Fe…Fe@FeS2表面,O2的O-O键断裂是自发的、无能垒的过程,最终生成了具有高自旋态(S=2)的FeIV=O物种;而在原始FeS2表面,此过程面临巨大的能垒,难以发生。
图3 材料表征:证实硫空位双Fe位点的成功构建
-
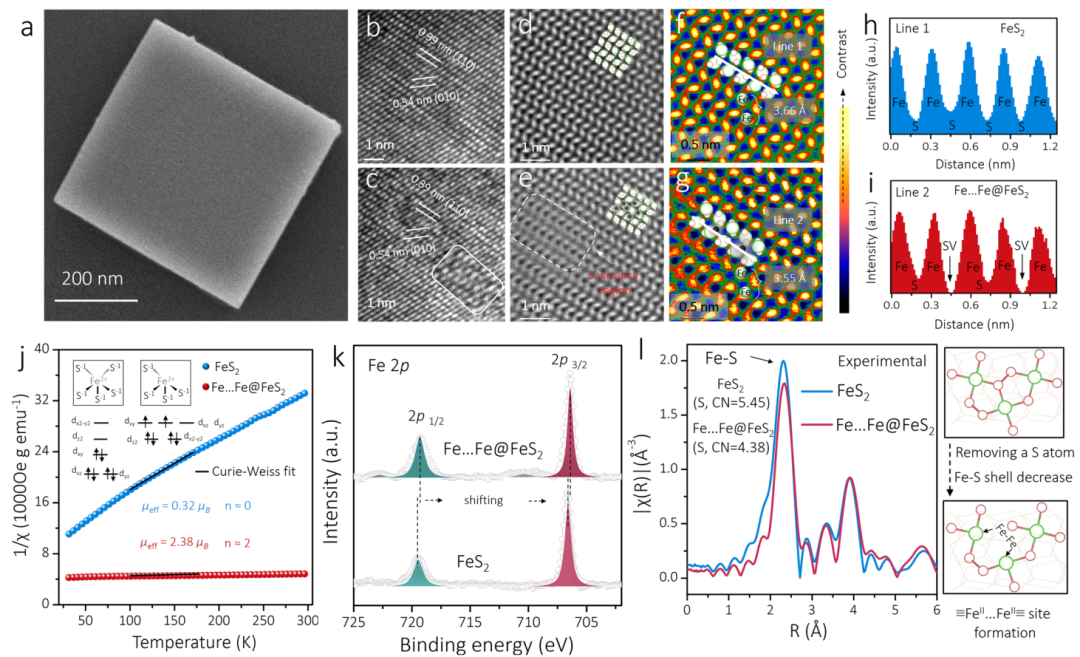
-
(a) (b) (c) SEM和HRTEM图表明,处理后的材料(Fe…Fe@FeS2)保持了原始FeS2的立方体形貌,但晶格条纹出现模糊,暗示了缺陷的形成。
(d)-(i) 原子级分辨率的HAADF-STEM图像和相应的强度曲线,从原子尺度上直接观察到了硫原子的缺失,并测量出双Fe位点的间距与理论模型高度吻合,为“双≡FeII…FeII≡位点”的成功构建提供了最直接的证据。
(j) 磁性测量结果从实验上证实了Fe的自旋态从未处理的低自旋(S=0)转变为处理后的中自旋(S=1)。
(k) XPS谱图显示Fe的结合能向低能端偏移,说明硫的移除导致电子在Fe位点上富集。
(l) X射线吸收谱(XAFS)分析表明,处理后Fe的平均配位数降低,与理论上从5配位向4配位的转变一致。
图4 机理研究:探究高自旋FeIV=O的生成与甲烷活化过程
-
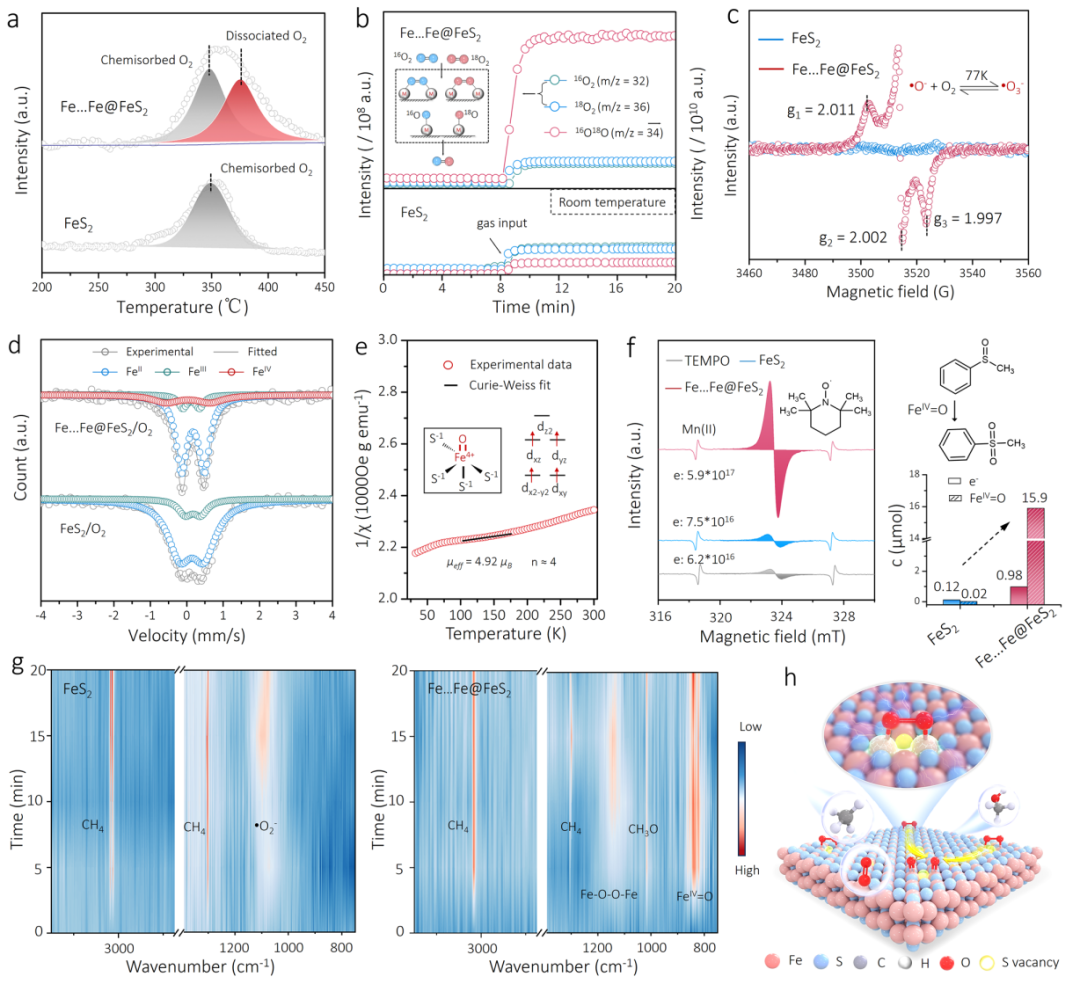
-
(a) O2程序升温脱附(TPD)谱图显示,Fe…Fe@FeS2上出现了新的对应于O2解离的信号峰。
(b) 氧同位素交换实验(16O2/18O2)表明,在Fe…Fe@FeS2表面,O-O键的断裂和重组非常迅速,证实了其优异的O2解离能力。
(d) 57Fe穆斯堡尔谱分析是本研究的关键证据之一,它直接检测到了O2处理后的Fe…Fe@FeS2样品中存在独特的FeIV物种,且其参数(同质异能位移和四极分裂)与高自旋(S=2)FeIV=O的特征完全吻合。
(e) 和 (f) 磁性测试和化学滴定进一步证实了高自旋FeIV=O物种的生成,并显示其可以被催化循环再生。
(g) 原位红外光谱(In-situ FTIR)在反应条件下“捕捉”到了关键的中间体:首先是理论预测的桥联≡Fe-O-O-Fe≡复合物和FeIV=O物种的振动峰,随后在引入CH4后,立即观察到了表面甲氧基(OCH3)的生成,清晰地描绘了反应的动态过程。
(h) 基于以上所有证据,绘制了清晰的反应机理示意图。
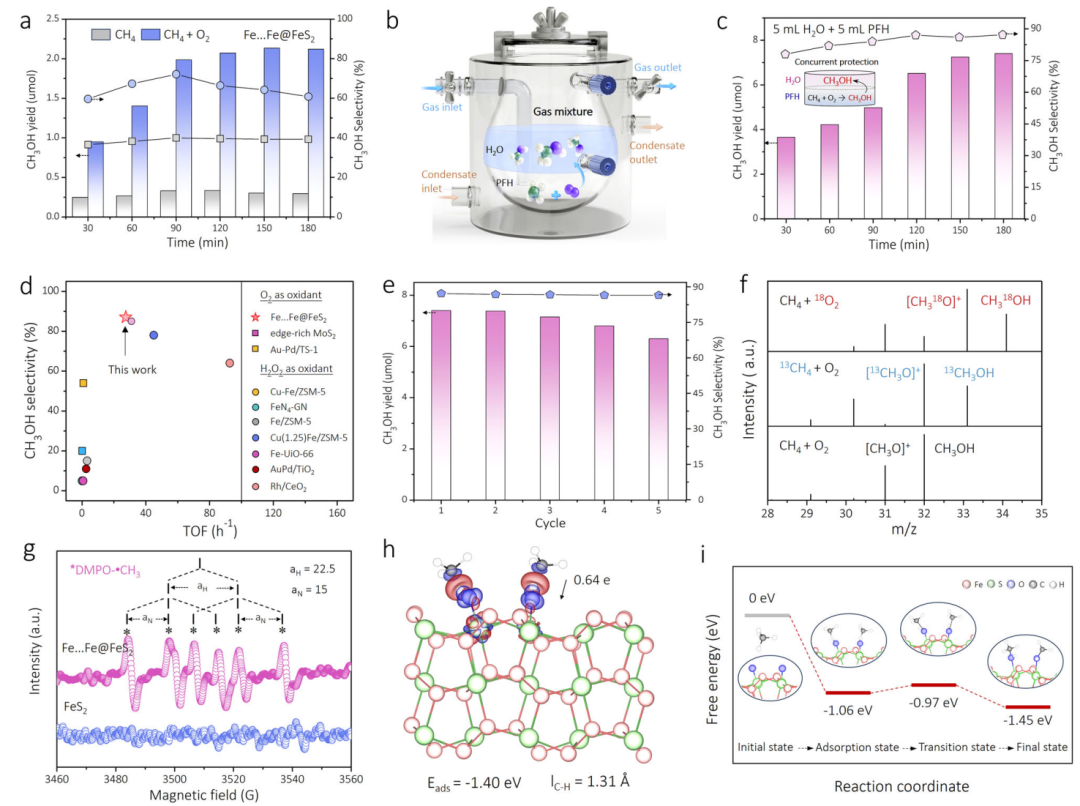
图5 催化性能:实现温和条件下甲烷向甲醇的高效选择性转化
-
-
(a) (b) (c) 性能测试表明,在常温常压下,Fe…Fe@FeS2能在O2存在下将CH4选择性地氧化为甲醇(CH3OH)。特别是在水/全氟己烷(PFH)双相体系中,由于PFH相能富集反应气体,而水相能及时萃取生成的甲醇防止其被过度氧化,反应的产率和选择性(87.0%)都得到了巨大提升。
(d) 催化活性的基准比较显示,该体系的转换频率(TOF=27.4 h-1)不仅优于大多数已报道的O2驱动体系,甚至超过了许多使用H2O2作氧化剂的体系。
(e) 循环实验证明了催化剂具有良好的稳定性。
(f) 同位素标记实验(13CH4和18O2)确凿地证明了产物甲醇中的碳原子来源于甲烷,氧原子来源于分子氧。
(g) (h) (i) 结合EPR捕捉到的甲基自由基(•CH3)和DFT计算,最终揭示了反应遵循经典的“氢提取-氧反弹”机理:高自旋FeIV=O物种以极低的能垒(0.09 eV)从CH4中夺取一个氢原子,随后生成的FeIII-OH与•CH3结合生成甲醇,并再生催化位点。
不得不说,这篇文章是一项构思极为巧妙且论证极为严谨的出色工作。它成功地将自然酶的智慧(双核铁中心激活O2)“嫁接”到了一种廉价的地球丰产矿物(黄铁矿)上,解决了非生物体系在温和条件下利用O2选择性氧化甲烷这一经典难题。
这项工作的核心亮点在于,作者没有停留在简单地“制造缺陷”,而是深刻理解并精准地利用了“缺陷工程”来“调控自旋态”。他们令人信服地揭示了一条清晰的逻辑链:移除桥联硫原子 → 改变Fe的配位环境和配体场 → 调控Fe的电子自旋态(从低自旋到中自旋) → 促进O2解离并生成高反应活性的高自旋FeIV=O → 实现高效的甲烷C-H键活化。每一个环节都由扎实的理论计算和多种先进的谱学表征(特别是原子级电镜、穆斯堡尔谱和原位红外)提供了确凿的证据。
这项发现的意义远不止于开发了一款优异的甲烷氧化催化剂。更重要的是,它为非均相催化领域提供了一种强有力的设计思想:通过对催化剂活性中心的几何与电子结构进行精修,特别是对金属的“自旋态”进行裁剪,可以从根本上调控其与反应物(如O2)的相互作用模式,从而突破反应的动力学瓶颈。这为理性设计用于其它惰性化学键(如C-C, N≡N)活化的高效催化剂打开了一扇新的大门。
文献引用:

1、凌灿灿:上海交通大学环境科学与工程学院博士后。研究方向为污染控制化学与环境纳米材料,致力于铁硫环境功能材料设计、自由基表界面生成与调控和有机物定向转化与资源化。主持上海市自然科学基金(青年)和中国博士后科学基金项目。以第一/通讯作者在Nat. Commun.、JACS(2篇)、Angew. Chem.、Sci. Bull.、Appl. Catal. B: Environ.等期刊发表论文7篇,其中1篇入选ESI高被引论文,授权国家发明专利1项。

2、李浩:上海交通大学环境科学与工程学院长聘教轨副教授,博士生导师,优秀青年科学基金项目(海外)获得者。从事污染控制化学、光/电催化和纳米环境材料等研究,负责和主持国家重点研发计划、国家自然科学基金、上海市科技创新计划等项目。以第一作者和通讯作者在Nat. Sustain.、Nat. Commun.、PNAS、JACS、Angew. Chem.等期刊发表论文40余篇,其中12篇入选ESI高被引论文,论文总引用8000余次,相关工作入选ES&T年度最佳论文和ACS Editors' Choice。目前担任Sustainability Science and Technology编委,Nano-Micro Letters、Fundamental Research青年编委,2019年获湖北省自然科学一等奖,2024年入选科睿唯安全球高被引科学家。

3、李美琪:上海交通大学环境科学与工程学院博士后,“博新计划”获得者。从事水污染控制、铁环境化学和纳米环境材料等领域的研究,主持国家自然科学基金(青年)、中国博士后科学基金、上海市“超级博士后”激励计划等项目。以第一/通讯作者身份在Nat. Commun. PNAS、JACS(2篇)、Angew、ES&T等期刊发表论文7篇,授权国家发明专利4项。

4、张礼知:国家杰出青年科学基金获得者,科技部中青年科技创新领军人才计划获得者,教育部长江学者特聘教授,中组部万人计划科技创新领军人才;现任上海交通大学上海交通大学特聘教授、环境科学与工程学院副院长,兼任中国可再生能源学会太阳光化学专业委员会委员、英国物理学会出版社旗下期刊Sustain. Sci. Technol.执行编委,化学学报、化学进展、环境化学、环境科学等杂志编委。主要从事污染控制化学、土壤/地下水污染修复、纳米环境材料制备及应用等研究,在Nat. Sustain.、Nat. Synth.、Sci. Adv.、Nat. Commun.、PNAS、JACS、Angew. Chem.等国际学术期刊发表论文420余篇,引用50000余次,H因子125,入选ESI高被引论文35篇,ESI热点论文1篇。获教育部高等学校科学研究优秀成果奖(科学技术)二等奖1项,湖北省自然科学一等奖1项和二等奖2项。连续入选科睿唯安全球高被引科学家、爱思唯尔中国高被引学者榜单。
转自https://mp.weixin.qq.com/s/YiEs9MhZvITxA-k0nuLrVQ