上海交通大学发表JACS:铑单原子配位环境调控促进反向氢溢流和电化学还原脱氯

第一作者:Qian Zheng; Hengyue Xu
通讯作者:Lizhi Zhang;Bo Yang;Yancai Yao
通讯单位:上海交通大学
论文DOI:10.1016/j.nanoen.2025.111619

成果简介
从二氧化钛表面氧到单原子催化中心的反向氢溢出(RHS)可以通过原子氢(H*)转移实现高效的电化学氢化,这一过程严重依赖于活性中心的配位环境和电子结构。在本研究中发现四氧配位Rh单原子电极(Rh1O4)在泡沫钛的水电解过程中表现出比五配位或三配位Rh单原子电极(Rh1O5或Rh1O3)更好的RHS性能。Rh−O配位数直接将Rh d带中心的相对位置调整到费米能级,从而调节了H+在Rh上的吸附以及二氧化钛表面氧与泡沫钛电极上Rh单原子之间的RHS效率.值得注意的是,四配位Rh1O4的优化氢吸附吉布斯自由能(ΔGH*)为+0.08 eV,接近配位氧原子的吸附自由能(−0.47 eV),大大降低了RHS势垒至+0.55 eV,明显低于Rh1O5和Rh1O3。这种结构优化表现出优异的电化学加氢性能,4-氯苯酚的降解速率常数为4.65 h−1,分别比Rh1O5(1.18 h−1)和Rh1O3(0.16 h−1)高4倍和29倍。本文的发现突出了单原子配位工程在定制原子级氢转移动力学方面的关键作用,并为设计可持续电化学加氢应用的高性能单原子电催化剂提供了战略框架。

图文摘要
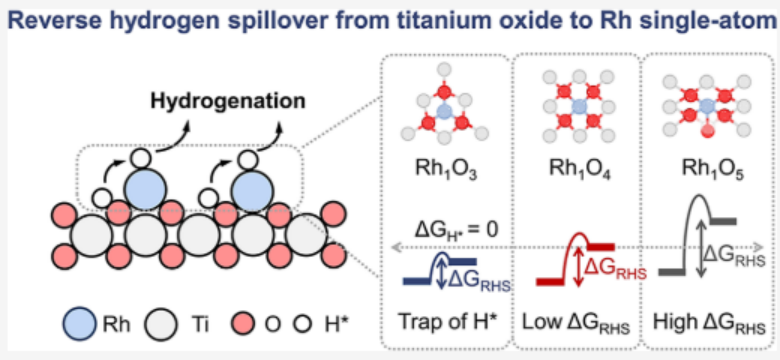

背景介绍
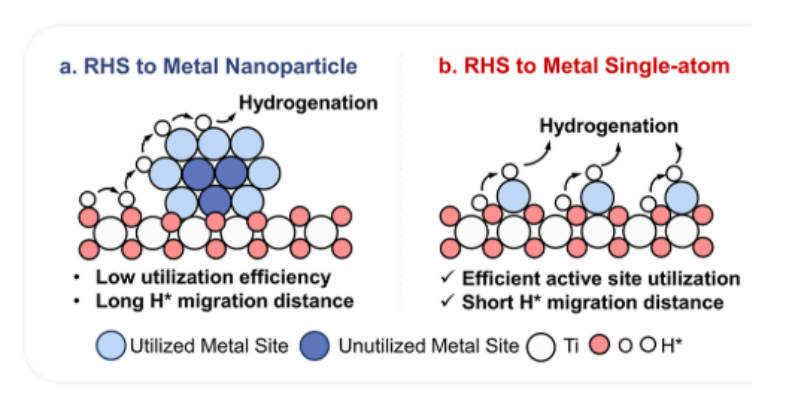
Figure 1. Schematic comparison of RHS to metal nanoparticle (a) and metal single-atom sites (b).
逆氢溢出(RHS)已成为电化学氢化中的一种关键现象,为能量存储、可持续化学合成和环境修复提供了转化潜力。传统电催化剂(RHSEs)通常由固定在可还原氧化物载体(如TiO2、WO3)上的金属纳米颗粒组成,其操作方式是在低过电位下产生原子氢(H*),并将其选择性地用于目标反应。然而,单原子RHSEs的出现代表了一种范式的转变,即H*从可还原载体迁移到孤立的金属位置。通过将氢溢出距离最小化到原子尺度(<1 nm),单原子RHSEs消除了纳米粒子系统固有的扩散瓶颈,实现了接近单位的H*利用效率(图1)。尽管有这样的期待,但这一领域仍然处于萌芽状态。特别是,单原子中心的配位几何构型(如M−O键数、键角)和电子结构(如分子轨道、氧化态)和它们的RHS活性之间的相互作用还没有被很好地理解。目前的研究缺乏原子尺度描述符(例如H*吸附能、溢出势垒)和宏观性能指标(例如加氢速率、法拉第效率)之间的系统相关性,从而阻碍了下一代RHSEs的合理设计。解开这些结构−活性关系对于解锁能够精确控制加氢的单原子催化剂至关重要,特别是对于生物质升级或污染物降解的复杂底物。
理论上,单原子RHS的发生需要H*从非金属配位原子(CAs,例如氧化物载体中的氧)有效地迁移到邻近的金属单原子(SA)位。这一过程由H*与SA/CA对之间的明显电子相互作用所支配,其中金属SA的离域d电子云限制了与H*的轨道重叠,形成弱的金属−H键,而非金属CA(例如,调节氧化态或配位环境)使氢吸附吉布斯自由能(ΔGH*)值理想地对齐SA和CA的吉布斯自由能(ΔGH*)来最小化这种能差,从而降低RHS过程中的氢吸附吉布斯自由能差,从而优化H*转移效率并实现高性能的单原子RHS电催化。
氢在催化剂表面的吸附吉布斯自由能受能带中心相对于费米能级的位置高度控制,与单原子位的配位环境和电子结构密切相关。为了阐明这种结构和活性关系,本文合成了Rh−O配位数可调的二氧化钛担载的Rh−O配位数(Rh1Ox,x=3,4和5)的Rh单原子催化剂。这使得系统地优化了氢在氧和Rh单原子(SA)位置上的吸附能,从而降低了RHS势垒,以驱动4-氯苯酚的高效电化学脱氯。通过结合密度泛函理论(DFT)模拟和现场电化学技术,本文探讨了单原子配位和电子结构如何调节能带中心排列、H吸附动力学和RHS效应,为催化机理提供了原子水平的见解。

图文导读
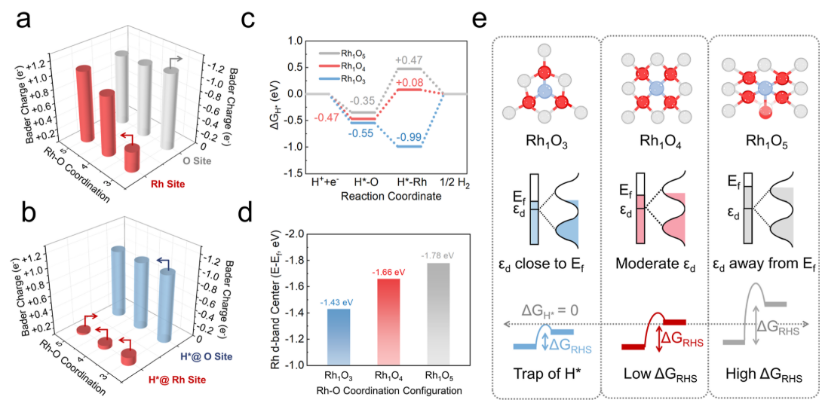
Figure 2. Theoretical insights into Rh single-atom RHS. (a,b) Bader charge analysis of Rh sites, coordinating O sites as well as H* adsorbed on Rh sites and O sites in Rh1O3, Rh1O4, and Rh1O5. (c) Hydrogen adsorption Gibbs free energy of H* adsorbed on Rh sites and O sites of Rh1O3, Rh1O4, and Rh1O5. (d) Rh d-band center of Rh1O3, Rh1O4, and Rh1O5. (e) Schematic illustration of RHS to single-atom sites with different d-band centers. Ef= Fermi level, εd = d-band center of metal single atom, ΔGRHS = Hydrogen adsorption Gibbs free energy differences in the RHS procedure.
本文首先通过Bader电荷分析(图2a)揭示了一个显著的趋势:在H*吸附之前,Rh−O配位从3增加到5,使Rh单原子的Bader电荷分别从+0.31 e−增加到+0.9 e−和+1.07 e−,而配位氧原子保持近恒定电荷(−1.06−1.13 e−)。这表明随着Rh−O配位数的增加,更多的电子从Rh单原子转移到二氧化钛衬底上。这种电子损失可以减弱Rh与H*之间的电子相互作用,这从吸附在Rh上的H*的Bader电荷减少所证实,从+0.12 e−(Rh1O3)转移到+0.06 e−(Rh1O4)和−0.03 e−(Rh1O5)(图2b)。这些发现建立了配位几何和电子转移之间的直接联系,突出了单个原子和H*之间的电子相互作用是调节原子RHS效率的关键杠杆。为了计算在Rh1Ox催化剂上的氢吸附动力学和RHS效率,本文计算了氧(O)和铑(Rh)中心的氢吸附吉布斯自由能(ΔGH*)(图2c)。对于Rh2O3,与O位(ΔGH*= −0.55 eV)相比,Rh位表现出较强的H捕获(ΔGH*= −0.99 eV),产生了阻碍H*脱附和随后参与加氢反应的热力学下沉。相反,Rh1O5具有明显的O的ΔGH*(−0.35 eV)和Rh的ΔGH*(+0.47 eV)的差异,导致高的能垒(ΔGRHS= +0.82 eV),阻碍了H*的迁移。值得注意的是,Rh1O4表现出最佳的平衡,因为Rh的近中性ΔGH*(+0.08 eV)阻止了H的捕获,同时与O位的中等ΔGH*(−0.47 eV)协同作用,共同将ΔGRHS降低到+0.55 eV。这种双重优化使Rh1O4中H*从O向Rh高效迁移,这可能是引发RHS现象的优良催化剂。
随后进行态密度分析,进一步阐明了Rh单原子在Rh1Ox上的发散H*吸附。 Rh的d带中心位置(εd)相对于费米能级从−1.43 eV(Rh1O3)逐渐下移到−1.66 eV(Rh1O4)和−1.78 eV(Rh1O5)(图2d),表明H*在Rh上的吸附依次减弱(Rh1O3>Rh1O4>Rh1O5),这与ΔGH*值的趋势很好地一致。这些结果表明,增加Rh−O配位减弱了Rh的d带电子密度,减少了它与H*的电子相互作用。关键的是,四配位的Rh1O4达到了最佳的εd位置(−1.66 eV),平衡了中等的H*亲和力(ΔGH*= +0.08 eV)和有效的氢化脱附,同时最小化了RHS势垒(ΔGRHS=+0.53 eV)。这种“金发姑娘”电子构型(图2e)确保了动态H*从氧转移到Rh而不捕获H,建立了依赖配位的带中心调制作为氢溢出过程的原子水平控制的通用杠杆。
合成与表征

Figure 3. Regulated coordination environment of single-atom Rh catalysts. (a−f) The coordination environment model, HAADF-STEM image, 3D intensity profile, and line intensity profile of Rh1O3 (a−c), Rh1O4 (d−f), and Rh1O5 (g−i). (j) XANES spectra and (k) Fourier transform (FT) at the Rh K-edge of Rh1O3, Rh1O4, Rh1O5, Rh2O3, and Rh foil. (l) X-ray photoelectron spectroscopy pattern of Rh1O3, Rh1O4, and Rh1O5 in the Rh 3d region.
本文对Rh2O3、Rh1O4和Rh1O5进行了高角度环形暗场扫描电子显微镜(HAADF-STEM)扫描。可以看到,图像中(图3b,e,h),孤立亮点的出现证实了Rh单原子的原子分散。原子分辨率Z对比分析,通过3D强度分布和线扫描(图3c,f,i),进一步验证了这种分散,具有对应于钛衬底上Rh的不同强度峰。进一步用X射线吸收精细结构(XAFS)和X射线光电子能谱(XPS)分析了Rh在Rh1Ox催化剂中的化学状态和局部配位(图3j−l)。位于Rh箔和Rh2O3参照物之间的Rh K边XANES谱(图3j)显示了中间氧化态(Rh0→Rh3+),这与配位工程中可调的电子结构一致。傅立叶变换EXAFS谱(图3k)在1.58 Å处显示一个主要的Rh−O散射峰,没有检测到Rh−Rh峰(∼2.42Å),证实了唯一的单原子分散。Rh 3d光谱(图3l)阐明了氧化态的过程,其中Rh1O3中的Rh0(307.5 eV)占主导地位,Rh1O4中的Rh0(307.5 eV)跃迁到Rh+(308.1 eV),而Rh1O5中的Rh3+(309.5 eV)与XANES的趋势一致,最终将配位环境与电子结构调制联系起来。
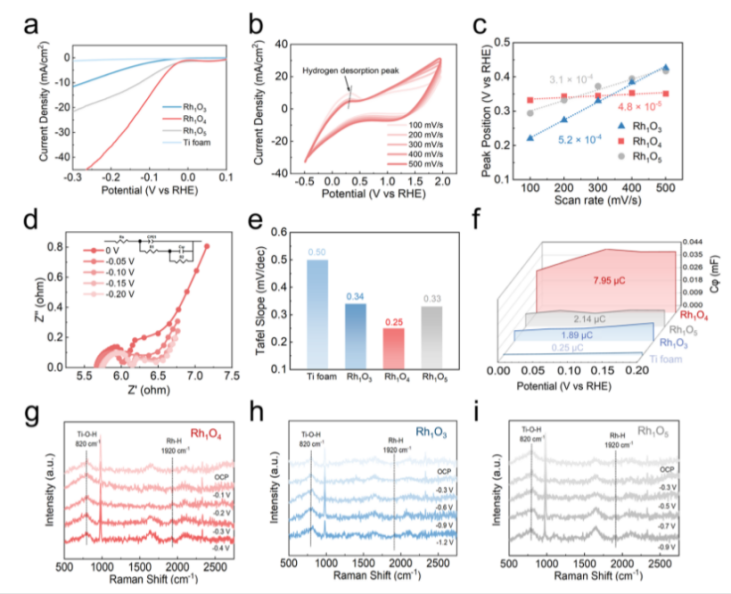
Figure 4. Experimental evidences for RHS on Rh1Ox. (a) LSV curve of Rh1O3, Rh1O4, and Rh1O5 and Ti foam in a N2 atmosphere. (b) CV curvesof Rh1O4with different scan rates. (c) Hydrogen desorption kinetics plots of Rh1O3, Rh1O4, and Rh1O5. (d) Nyquist plots of Rh1O4. Inset:equivalent circuit for the EIS simulation. (e) EIS-derived Tafel slope of Rh1O3, Rh1O4, and Rh1O5 and Ti foam. (f) QH* plots of Rh1O3, Rh1O4, andRh1O5and titanium oxide substrate acquired from EIS simulation. (g−i) In situ Raman analysis of Rh1O4, Rh1O3, and Rh1O5.
为了探索缺陷对二氧化钛底物催化性能的潜在影响,本文用线性扫描伏安法(LSV)结合电子顺磁共振(EPR)和电化学阻抗谱(EIS)研究了水解离活性、电导率以及Rh1Ox催化剂和相应的泡沫钛衬底的H*生产能力。LSV曲线(图4a)显示,Rh1O4在显著正的起始电位(−0.07 V vs RHE)下获得了10 mA cm−2的电流密度,而Rh1O3(−0.26 V vs RHE)和Rh1O5(−0.13 V vs RHE) 具有明显的正起始电位。而在没有Rh负载的情况下,裸泡沫钛基板的起始电势表现出与−1.038 V∼−0.966 V相当的起始电位,缺陷的存在使裸泡沫钛基板的电流密度略有增加。然而,这种改善是不能与Rh单原子的引入相比的,这表明与Rh单原子的RHS效应相比,Ti3+和氧空位在H*脱附中的作用可以忽略不计。
此外通过在循环伏安(CV)曲线上检测不同扫描速率(100−500 mV s−1;图4b)中的氢脱附峰,通过绘制峰电位与扫描速率的关系图(图4c)计算出的曲线斜率显示5.2×10−4s(Rh1O3),3.1x10−4 s (Rh1O5)和4.8×10−5 s(Rh1O4),其中Rh1O4的最小斜率表示H*解吸和RHS动力学的加速。这些结果明确地证实了Rh1O4是动态H*运输的最佳催化剂,这与配位调制d带定位的理论预测一致。
因此,本文利用交流阻抗谱进一步阐明了氢的吸附动力学和RHS动力学。用双路径等效电路模型拟合Nyquist图(图4d)显示电荷转移电阻,由此计算出EIS导出的Tafel斜率(图4e)。Rh1O4表现出最小的塔菲尔斜率(0.25 mV dec−1),优于Rh1O3(0.34 mV dec−1)、Rh1O5(0.33 mV dec−1)和裸泡沫钛(0.50 mV dec−1),验证了其优越的吸氢动力学和RHS效率。作为补充,氢吸附电荷(QH*)通过积分依赖于电位的伪电容(Cφ)得到,量化了表面H*分布。负载Rh单原子使QH*从0.25 μC(裸钛)增加到1.89 μC(Rh2O3),证实了Rh作为H*吸附中心的作用(图4f)。值得注意的是,Rh1O4 的QH*达到了7.95 μC的4.2倍,表明H*通过RHS有效地从TiO2位迁移到Rh位。相反,Rh1O5 的QH*表现出剧烈的下降,降至2.14μC,与Rh1O3相似。这些独特的电化学表征与DFT预测的d带定位相结合,最终确定Rh1O4是RHS驱动的H*传输的最佳几何构型。
然后用原位拉曼光谱研究了在不同外加电位(图4g−i)下,在磷酸盐缓冲溶液(PBS)中,Rh1Ox催化剂对H*的吸附动力学。Rh−H伸缩振动(1920 cm−1)表明H*在Rh单原子上的吸附出现在截然不同的电位下:−1.2 V(Rh1O3),−0.1 V(Rh1O4),和−0.5 V(Rh1O5)。Rh1O4需要最小的负偏压来激活Rh−H键的这一事实直接证实了其优化的Rh动力学,使H*在最温和的条件下从二氧化钛迁移到Rh。光谱证据与密度泛函和电化学数据相吻合,通过最佳的d带位置和较低的能垒,巩固了四配位的Rh1O4作为RHS介体的优势。
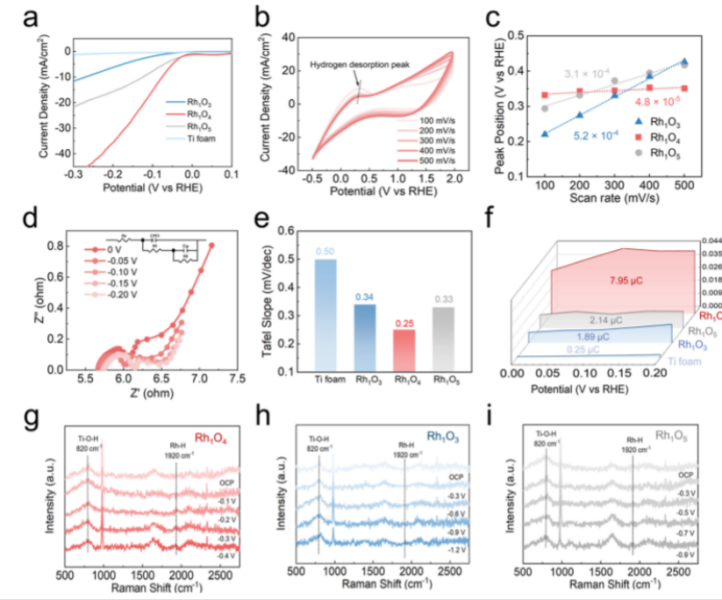
Figure 5. 4-CP degradation performance and mechanism investigation. (a) Schematic illustration of H* generation, transfer, and 4-CP hydrogenation process. (b) 4-CP degradation on Rh1O3, Rh1O4, Rh1O5, and Ti foam. (c) Faradaic efficiency of 4-CP hydrogenation on Rh1O3, Rh1O4, and Rh1O5. (d) Stability experiment for 4-CP hydrogenation on Rh1O4 in 20 rounds of electrolysis. (e) 4-CP degradation on Rh1O3, Rh1O4, and Rh1O5 with 0.1 mol/L of TBA. (f) EPR spectra. (g) KIE plot of 4-CP degradation rate constant ratio (KIE = kH2O/kD2O). h. Schematic illustration of Rh−O coordination-derived differences in single-atom RHS capacity and 4-CP hydrogenation activity.
鉴于Rh1Ox催化剂的RHS容量得到增强,本文以对氯苯酚(4-CP)为模型污染物对其电化学加氢性能进行了评估(图5a)。在−0.6 V vs Hg/HgO(−0.09 vs RHE)的温和应用电位下,Rh1O4在45min内可实现99%的4-CP脱氯率(图5b),显著优于Rh1O3(1h内18%)和Rh1O5(1h内72%)。动力学分析表明,Rh1O4的最高速率常数为4.65 h−1,分别是Rh1O5(1.18 h−1)和Rh1O3(0.16 h−1)的4倍和29倍,并超过大多数基准电催化剂。这种性能梯度以及Rh1O4的优异法拉第效率(图5c)与RHS容量直接相关,证实了从二氧化钛到Rh位的优化H*传输最大限度地利用了目标C−Cl键的断裂。随后连续20多个循环的稳定性测试表明,4-CP的去除率一直超过95%,没有显著的性能下降(图5d)。
为了阐明原子氢(H*)在4-CP加氢中的中心作用,本文以0.1M叔丁醇为H*清除剂进行了H*猝灭实验。叔丁醇的引入使Rh1O4的4-CP降解速率常数从4.65 h−1降低到1.15 h−1,与Rh1O3(25%,0.16 h−1→0.12 h−1)和Rh1O5(44%,1.18 h−1→0.66 h−1)的降幅较小形成鲜明对比(图5e)。Rh1O4对H*抑制的这种明显的敏感性突出了它对RHS介导的H*转移催化活性的独特依赖,验证了RHS效率在驱动高效的C−Cl键断裂中的关键作用。采用电子顺磁共振结合DMPO捕集的方法研究了4-CP加氢过程中H*的利用动力学。在引入4-CP之前,Rh1O4表现出最强的DMPO-H信号强度(Rh1O4>Rh1O5>Rh1O3>钛泡沫;图5f),与其优越的H*生成(LSV,图4a)和吸附容量(QH*,图4f)一致。
最后进行了动力学同位素效应(KIE)分析,以评估H*转移在4-CP氢化速率决定步骤中的作用。根据水和D2O中的反应速率(图5g)得出的KIE值遵循如下顺序:Rh1O3(1.48)>Rh1O5(1.33)>Rh1O4(1.20)。Rh1O4的近单位KIE(<1.3)表明其对H*迁移率的动力学阻力最小,这与其优化的RHS动力学一致。这与Rh2O3和Rh2O5不同,它们较高的KIE值反映了由于Rh−O配位不佳而造成的H*运输限制。这些结果明确地将RHS效率与加氢活性相关联,证实了四个氧配位的Rh1O4离子降低了H*迁移的能垒,从而加速了H*介导的4-CP脱氯过程(图5h)。
结果展望
综上,本文建立了单原子配位环境与RHS过程中H*吸附、迁移、解吸和氢化的原子水平动力学之间的明确关联。对于泡沫钛负载的Rh单原子电催化剂,Rh−O配位调节Rh d带中心相对于费米能级的位置,从而调节H*在Rh位的吸附以及二氧化钛表面氧与单原子Rh位之间的RHS效率.四配位Rh单原子电极实现了战略位置的d带中心(−1.66 eV vs Fermi Level,DFT),优化了H*吸附能(+0.08 eV),同时促进了氢气的脱附,并降低了Rh1O5的势垒(+0.55 eV,vs +0.82 eV)。这种配位驱动的电子调谐使四个氧配位的Rh1O4电极的性能超过了五个或三个配位的电极,在4-CP加氢中分别提高了4倍和29倍。本文的工作阐明了配位工程在掌握RHS动力学中的关键作用,为单原子电催化剂在节能加氢等方面提供了通用的设计原则。
转自https://mp.weixin.qq.com/s/Pwt9Q0d1dhpgv2f1eFP46Q