华中师大张礼知等:表面硫酸根修饰促进BiOBr激子提取及单线态氧生成用于氯酚脱氯

表面硫酸根修饰促进溴氧化铋纳米片激子提取,实现高效单线态氧光合成和氯酚脱氯
石彦彪,杨治平,张陈陈,陈嫔吉,傅浩洋,刘旭鹏,李浩,赖波,张礼知
上海交通大学,环境科学与工程学院
四川大学,建筑与环境学院
华中师范大学,化学学院
成都工业学院,材料与环境工程学院
南洋理工大学,材料科学与工程学院
【文献链接】
【背景介绍】
光催化分子氧(O2)活化为有机污染物解毒提供了一种可持续生成单线态氧(1O2)途径。然而,传统载流子参与的路径常因光生空穴介导的超氧自由基(•O2⁻)氧化而导致能量损耗,影响了1O2产率效率。若能将光生活性物种在其完全分离成为电子-空穴对前直接提取至光催化剂表面,则可规避传统内建电场驱动的载流子提取过程,促进电中性激子介导的1O2合成。本研究通过简便的光刻蚀-配位策略,在溴氧化铋纳米片(BiOBr)表面引入硫酸根离子([SO4]-BiOBr),调控其空间位阻及表面激子态。得益于体相与表面激子态形成的能量梯度,[SO4]-BiOBr展现出极高的体相激子提取效率。不同于对照样品BiOBr通过“side-on”模式促进化学态O2吸附,具有较大空间位阻的[SO4]-BiOBr能够优先将长寿命激子的能量转移至物理吸附态O2,导致光催化产生的活性氧物种从•O2⁻转变为1O2,从而显著提升4-氯苯酚(4-CP)的脱氯降解效率。该研究揭示了半导体材料表面修饰对其激子过程调控的关键作用,为高效1O2光合成和后续废水净化提供了新思路。

【文章亮点】
1. 研究通过光蚀刻-配位策略在BiOBr纳米片表面引入硫酸根([SO4]),构建了从体相到表面的激子态能量梯度,实现高效体相激子提取,显著提升1O2光合作用效率;
2. [SO4]修饰增大了BiOBr表面空间位阻,促使O2以物理吸附模式(而非化学吸附)与催化剂表面结合,通过能量转移路径而非传统电荷载流子路径选择性生成1O2,避免•O2⁻等副产物产生;
3. [SO4]-BiOBr在可见光下表现出优异的氯酚类污染物(如4-氯酚)脱氯和降解性能,脱氯率超过96%,且具备良好稳定性和环境适应性,为有机废水净化提供了新策略。
【内容简介】
日前,华中师范大学张礼知教授、四川大学赖波教授和上海交通大学李浩副教授在Rare Metals上发表了题为“Surface sulfate modification promoted excitons extraction of BiOBr nanosheets for efficient 1O2 photosynthesis and chlorophenols dechlorination”的研究文章,提出了光刻蚀-配位策略在BiOBr纳米片表面引入硫酸根离子([SO4]-BiOBr),促使光催化产生的活性氧物种由•O2⁻转变为1O2,显著提升了4-氯苯酚(4-CP)脱氯和降解效率。
本文针对光催化O2活化生成1O2过程中载流子参与路径效率低、选择性差的问题,提出利用硫酸根修饰调控BiOBr纳米片表面空间位阻及激子态。研究发现,[SO4]修饰不仅建立了体相至表面激子态的能量梯度,促进体相激子提取,还通过位阻效应诱导催化剂表面化学态吸附的O2转变化物理吸附模式,从而通过直接能量转移过程生成1O2。该策略显著提升了光化学合成1O2的选择性(89.5%)和氯酚类污染物的脱氯效率,为氯酚类废水处理提供了新思路。
【图文解析】

图1. 结构表征。BiOBr和[SO4]-BiOBr的(A)理论模型、(B)XRD图谱和(C)拉曼光谱。(D) Na2SO3溶液中光蚀刻后未洗涤和洗涤后[SO4]-BiOBr样品的高分辨率S 2p XPS谱。(E, F) [SO4]-BiOBr的TEM和HRTEM图。(G-J) [SO4]-BiOBr的STEM-EDX mapping图像(Bi, S和Br)。
XRD谱图表明,表面[SO4]修饰并未改变BiOBr的特征衍射峰(JCPDS# 73-2061),只是降低了整体峰强度。拉曼光谱表明,在Na2SO3溶液中光蚀刻过程改变了BiOBr表面原子排列。在S 2p XPS图谱中,168.1 eV处出现的信号峰表明光蚀刻-交换过程在BiOBr表面引入了[SO4]物种。这种表面[SO4]修饰不会改变BiOBr纳米片的主要暴露晶面,只会引起局部原子结构畸变。EDX mapping显示S元素均匀分布在[SO4]-BiOBr表面。

图2. 表面化学性质。(A) BiOBr-OV和[SO4]-BiOBr表面OV形成能垒。(B) BiOBr, BiOBr-OV和[SO4]-BiOBr的EPR谱。高分辨率XPS谱:(C) Bi 4f, (D) O 1s 和 (E) Br 3d。
原始BiOBr表面引入OV需要克服较高能垒(4.12 eV),而其在Na2SO3溶液中可见光照射下会转变为热力学自发过程(-2.40 eV)。更重要的是,[SO3]与BiOBr表面晶格氧反应原位生成的[SO4]可以通过强S-O-Bi化学键吸附在材料表面(具有较大脱附能垒2.92 eV),而非弱物理吸附。EPR谱图表明,[SO4]-BiOBr在g=2.003处OV信号强度明显高于BiOBr-OV),证实了表面[SO4]修饰能够诱导BiOBr表面生成OV。与BiOBr相比,[SO4]的O原子与BiOBr表面强结合导致Bi 4f、O 1s和Br 3d结合能降低。
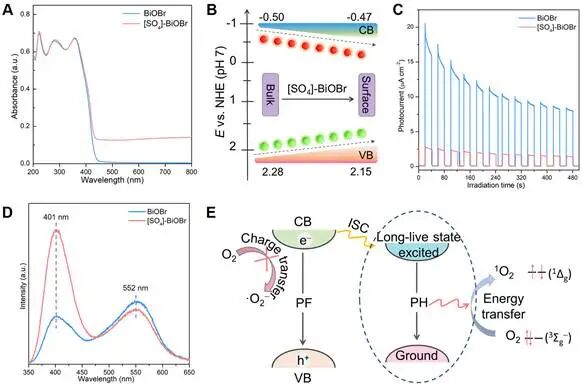
图3. 电荷载流子动力学表征。(A) BiOBr和[SO4]-BiOBr的UV-vis DRS。(B) [SO4]-BiOBr从体相到表面的空间能带排列示意图。(C) BiOBr和[SO4]-BiOBr的瞬态光电流和(D)室温稳态PL光谱。(E) 能量转移途径增强¹O2生成示意图。
UV-vis光谱表明表面[SO4]修饰几乎不改变BiOBr的光吸收范围,但在可见光范围内出现明显拖尾。与原始BiOBr相比,表面[SO4]修饰导致材料的导带(CB)电位从-0.50略微增加到-0.47 eV,而价带(VB)电位从2.28降低至2.15 eV。由此可知,[SO4]-BiOBr表面与体相之间的CB和VB电位能级差分别为30和130 meV,这种空间能量梯度能够驱动光生物种从材料体相迁移至表面。瞬态光电流测试表明[SO4]-BiOBr比原始BiOBr具有更稳定且更小的电流密度。比BiOBr相比,[SO4]-BiOBr的阻抗图谱的圆弧半径更大,表明[SO4]修饰增加了界面电荷转移阻力。在室温下,BiOBr具有带间发射(401 nm)和缺陷态带内发射(552 nm)信号,而表面[SO4]修饰导致缺陷态诱导发射减弱、固有的带间发射增强,意味着[SO4]-BiOBr的激子效应增强。鉴于荧光(PL)通常源于自旋允许的辐射衰变,而磷光(PH)是由自旋禁阻的长寿命态辐射衰变引起,由此可推断出表面[SO4]修饰很大程度上抑制了BiOBr的电子转移过程,促进[SO4]-BiOBr中产生大量长寿命的束缚态激子。

图4. 光催化O2活化和ROSs鉴定. (A) TEMP-1O2加合物EPR谱,(B) DMPO-•O2⁻和(C) BiOBr, BiOBr-OV, [SO4]-BiOBr和(BiOBr+[ SO4])累积的1O2产率。(D) 以[SO4]-BiOBr为光催化剂,在不同溶剂中FFA的降解曲线。(E) [SO4]-BiOBr中时间分辨的•O2⁻、•OH和1O2浓度。(F) BiOBr, BiOBr-OV, [SO4]-BiOBr和(BiOBr+[SO4])中累积ROS浓度和1O2与•O2⁻的比率。(G) BiOBr, BiOBr-OV, [SO4]-BiOBr和(BiOBr+[SO4])产生的非自由基与自由基的比率。
以TEMP作为1O2捕获剂,ESR测试表明[SO4]-BiOBr显示出远强于原始BiOBr的三重峰信号(1:1:1, aN= 16.9 G),表明生成了大量1O2。如前所述,1O2可以直接由能量转移主导的O2活化途径产生,也可以间接由光生空穴介导的•O2⁻氧化反应产生。鉴于BiOBr-OV的TEMP-1O2信号显著降低,排除了表面OV介导的1O2生成路径。相反,使用DMPO捕获•O2⁻时,BiOBr-OV样品中观察到显著增强的DMPO-•O2⁻信号(aN=12.9 G, aH=10.3 G)。由此可知,产生的1O2来源于能量转移途径参与的O2活化过程,而非电荷载流子介导的途径。以糠醇(FFA)作为化学探针定量分析1O2产量,我们发现[SO4]-BiOBr的累积1O2产率随着O2浓度的增加而逐渐升高,这可能是因为充足的O2供应抵消了传质限制。同时,[SO4]-BiOBr的1O2产生浓度高达59.45 μmol L-1,远超BiOBr(16.45 μmol L-1)、BiOBr-OV(1.68 μmol L-1)和[SO4]+BiOBr(24.17 μmol L-1)。为了消除光生空穴和ROS直接氧化FFA的干扰,我们在1O2寿命更长的氘代水(D2O)中评估了FFA的氧化效率。随着D2O浓度增加,FFA的降解速率逐渐增加,证实了1O2确实是氧化FFA的物种。我们还通过监测光催化O2活化过程中其它ROS(•O2⁻和•OH)的产率来评估1O2选择性,发现[SO4]-BiOBr产生的1O2浓度超过了•O2⁻和•OH。与BiOBr、BiOBr-OV和[SO4]+BiOBr相比,BiOBr表面引入[SO4]可以极大提高1O2和•O2⁻的浓度比(15.75)。由此可知,[SO4]-BiOBr具有更高的1O2生成选择性(89.5%),即其能够通过能量转移途径高效活化O2产生1O2。
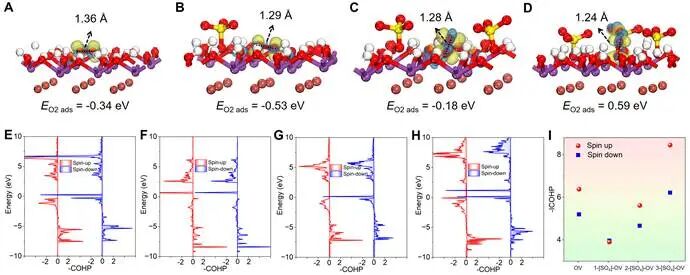
图5. O₂吸附在光催化剂表面的构型、能量以及晶体轨道哈密顿布居(COHP)图:(A, E) BiOBr-OV, (B, F) 1-[SO4]-BiOBr, (C, G) 2-[SO4]-BiOBr和 (D, H) 3-[SO4]-BiOBr。(I) O2吸附在BiOBr-OV和x-[SO4]-BiOBr上相应的ICOHP值。
无缺陷的BiOBr(001)表面无法提供有效吸附和活化O2的位点,而引入氧空位可以很好地解决该难题。对于BiOBr-OV,其(001)表面的空间位阻相对较小,O2的两个氧原子会以侧向桥接模式在OV处相互作用,从而拉伸O-O键长至1.36 Å,接近•O2⁻的键长(1.26 Å)。有趣的是,在OV附近引入[SO4]后O-O键拉伸程度变小(1.28 Å),进一步增加OV附近[SO4]的浓度对O2的O-O键拉伸影响更小(O2分子的O-O键为1.21 Å),说明空间位阻增加阻碍了材料与O2的化学相互作用。为了研究O2在不同样品表面的成键/反键特性,我们计算了O2吸附在BiOBr-OV和x-[SO4]-BiOBr(x=1,2,3)表面的晶体轨道哈密顿布居(COHP)。由于O2吸附在1-[SO4]-BiOBr表面的O-O键强度远弱于其他材料,表明其化学键更易活化乃至断裂。BiOBr表面更高的[SO4]浓度导致O-O键强度增加,阻碍了其化学键的拉伸和活化。因此,我们从理论上证明[SO4]修饰将增加BiOBr的表面空间位阻,导致O2由强化学吸附模式转变为弱物理吸附,并通过抑制电荷载流子途径进一步促进能量转移介导的O2活化生成1O2。

图6. 光催化4-CP降解. (A) 4-CP的光催化降解曲线和(B) BiOBr, BiOBr-OV, [SO4]-BiOBr和 (BiOBr+[SO4]) 相应的反应速率常数。(C) BiOBr, BiOBr-OV, [SO4]-BiOBr和 (BiOBr+[SO4])中4-CP的时间分辨脱氯率。(D) [SO4]-BiOBr中4-CP的长期光催化降解性能。(E) [SO4]-BiOBr中4-CP的可能降解路径。(F) BiOBr和[SO4]-BiOBr光降解不同氯酚污染物的反应速率常数。
氯酚是合成农药、药物和染料的关键中间体,其强生物毒性的氯原子引发了严重的环境安全问题。鉴于具有强激子效应的二维光催化剂可以通过能量转移参与的O2活化过程高效产生1O2,其能够选择性攻击4-CP的氯原子。与原始BiOBr相比,[SO4]-BiOBr在可见光下具有最高的4-CP降解活性,其表观反应速率常数为2.76 ×10-2 min-1,是原始BiOBr(0.45×10-2min-1)的6.1倍。尽管已有工作报道光催化剂表面引入OV可以将O2优先活化为•O2⁻,促进氯酚脱氯和苯环开环。然而,具有较高•O2⁻积累浓度的BiOBr-OV仅实现了低于60%的脱氯率和37.6%的TOC去除率,而[SO4]-BiOBr可在120分钟内有效降解4-CP,具有更高的脱氯率(96%)。这些结果一致表明具有高效1O2合成能力的[SO4]-BiOBr在氯酚废水处理方面具有显著优势。我们进一步分析了材料的4-CP脱氯和降解路径:1O2会首先攻击4-CP分子,导致苯环结构上氢原子被酮基取代形成1,4-苯醌,1,4-苯醌可被1O2进一步分解形成小分子有机酸(如马来酸、草酸和乙酸),并最终转化为CO2和H2O。为了进一步检验1O2介导的脱氯和降解途径是否适用于其他氯代有机污染物,我们选择了五种典型的氯酚化合物(对氯酚、2,4-二氯酚、2,4,5-三氯酚、2,3,4,6-四氯酚和五氯酚)作为模型污染物。与原始BiOBr相比,[SO4]-BiOBr对氯酚类化合物均表现出更高的降解速率,证明具有高选择性产生1O2的[SO4]-BiOBr在氯酚类污染物脱氯和降解过程中具有巨大优势。
【全文小结】
1.[SO4]基团修饰增加了BiOBr纳米片表面空间位阻,促使O2从易于发生电荷转移的侧向桥连化学吸附模式转变为弱物理吸附,抑制了电荷载流子参与的超氧阴离子(•O2⁻)生成;
2. [SO4]-BiOBr的长寿命激子能够直接将能量传递至弱物理吸附态O2,从而将活性氧物种从•O2⁻调控为1O2,显著提高了1O2产率(59.45 μmol·L-1)和选择性(89.5%);
3.具有高效产生1O2能力的[SO4]-BiOBr展现出优异的4-氯酚(4-CP)降解活性和脱氯效率(>96%),且能够有效降解多种氯酚和其它有机污染物,证明了其在难降解有机废水净化中的应用潜力。
【作者简介】

石彦彪,硕导,主要从事环境小分子(O2和CO2)活化和工业含氮废水处理研究。入选天府峨眉青年人才(2024)、四川大学“双百人才计划”B类。以第一/共同通讯作者在Chem、Nat. Commun.、Angew. Chem. Int. Ed.、Adv. Mater.、Environ. Sci. Technol.、Appl. Catal. B-Environ.等化学、材料及环境领域顶级期刊发表学术论文50余篇,包括5篇Nature Index收录期刊论文,其中4篇入选ESI高被引用论文,当前H指数为31。目前担任Rare Metals、Exploration和Chinese Chemical Letters期刊青年编委。

张礼知,教授,博导,国家杰出青年科学基金获得者,科技部中青年科技创新领军人才计划获得者,教育部长江学者特聘教授,中组部万人计划科技创新领军人才;现任上海交通大学上海交通大学特聘教授、环境科学与工程学院副院长,兼任中国可再生能源学会太阳光化学专业委员会委员、英国物理学会出版社旗下期刊Sustain. Sci. Technol.执行编委,化学学报、化学进展、环境化学、环境科学等杂志编委。主要从事污染控制化学、土壤/地下水污染修复、纳米环境材料制备及应用等研究,在Nat. Sustain.、Nat. Synth.、Sci. Adv.、Nat. Commun.、PNAS、JACS、Angew. Chem.等国际学术期刊发表论文420余篇,引用50000余次,H因子125,入选ESI高被引论文35篇,ESI热点论文1篇。获教育部高等学校科学研究优秀成果奖(科学技术)二等奖1项,湖北省自然科学一等奖1项和二等奖2项。连续入选科睿唯安全球高被引科学家、爱思唯尔中国高被引学者榜单。

赖波,教授,博导,环境科学与工程系主任,长期从事高级氧化处理技术理论与装备的研究。入选国家重大人才工程特聘教授(2024)、国家人才计划青年学者(2020)、天府青城科技创新领军人才(2024)、Clarivate全球高被引学者(2022)、爱思唯尔中国高被引学者(2022-2024)。以第一作/通讯作者在Nat. Commun.、Angew. Chem. Int. Ed.、ACS Catal.、Environ. Sci. & Technol.、Water Res.等主流环境期刊发表SCI论文200余篇;授权中国专利26件,国际专利(美国)2件,专利转让许可14件,成果已应用于废水处理工程多项。关键技术入选2022年《国家先进污染防治技术目录(水污染防治领域)》,获四川省科技进步一等奖1项(1/10,2020)、环境保护科学技术发明二等奖(1/6,2024)、中国环保产业协会环境技术进步一等奖2项(1/15, 2020;2/15, 2022)、第二十四届中国专利优秀奖1项、中国环境科学学会青年科学家奖(金奖)、四川省青年科技奖、第一届全国博士后创新创业大赛银奖(创业组)、第二十四届中国科协求是杰出青年成果转化奖提名奖。担任中国城市科学研究会水环境与水生态分会副秘书长,Chinese Chemical Letters执行主编、Environment Functional Materials副主编(创刊);带领团队培养学生获得国家自然科学基金博士生项目2项,全国性奖学金12人次,国家奖学金30余人次,中国国际大学生创新大赛银奖1项,省级竞赛金奖4项,四川大学“十佳”研究生2人,四川大学学术之星3人次。

李浩,博导,上海交通大学环境科学与工程学院长聘教轨副教授,博士生导师,优秀青年科学基金项目(海外)获得者。从事污染控制化学、光/电催化和纳米环境材料等研究,负责和主持国家重点研发计划、国家自然科学基金、上海市科技创新计划等项目。以第一作者和通讯作者在Nat. Sustain.、Nat. Commun.、PNAS、JACS、Angew. Chem.等期刊发表论文40余篇,其中12篇入选ESI高被引论文,论文总引用8000余次,相关工作入选ES&T年度最佳论文和ACS Editors' Choice。目前担任Sustainability Science and Technology编委,Nano-Micro Letters、Fundamental Research青年编委,2019年获湖北省自然科学一等奖,2024年入选科睿唯安全球高被引科学家。
转自https://mp.weixin.qq.com/s/vqKjtHrEaMxRn4XFc8VOYg